Translate this page into:
Novel mutation in the nuclear receptor subfamily 0, group B, member 1 (NR0B1) gene associated with intrafamilial heterogeneity in three boys with X-linked adrenal hypoplasia congenita and hypogonadotropic hypogonadism from India
2 Department of Child Health, Christian Medical College and Hospital, Vellore 632004, Tamil Nadu, India
Corresponding Author:
Sumita Danda
Department of Medical Genetics, Christian Medical College and Hospital, Vellore 632004, Tamil Nadu
India
sdanda@cmcvellore.ac.in
| How to cite this article: Mohan S, Danda S, Mathai S, Simon A. Novel mutation in the nuclear receptor subfamily 0, group B, member 1 (NR0B1) gene associated with intrafamilial heterogeneity in three boys with X-linked adrenal hypoplasia congenita and hypogonadotropic hypogonadism from India. Natl Med J India 2019;32:141-143 |
Abstract
Background. Nuclear receptor subfamily 0, group B, member 1 (NR0B1) gene previously known as DAX1 is a transcription factor that plays a key role in the development of hypothalamo-pituitary-gonadal and adrenal axis. Primary adrenal failure may result from metabolic, infection, autoimmune or developmental causes resulting in a life-threatening condition needing immediate intervention. This study aimed to analyse NR0B1 (DAX1) gene mutation resulting in adrenal hypoplasia congenita (AHC) in three brothers presenting with hypogonadotropic hypogonadism and primary adrenal failure either in infancy or in early childhood.Methods. We studied three boys with primary adrenal failure and hypogonadotropic hypogonadism presenting at different ages at the Paediatric Endocrinology Clinic. Muta- tional analysis of NR0B1 gene was carried out by bidirectional sequencing.
Results. All the three boys had deletion of G in exon 1 at position 189 (c.189_189delG) of the gene resulting in frame shift mutation (Y64Tfs*21).
Conclusion. Novel mutation in NR0B1 detected by this study explained the cause ofhypogonadotropic hypogonadism with primary adrenal failure in this Indian family. Intrafamilial variability was seen in this family. Early diagnosis by genetic testing, genetic counselling and family screening can help to manage this life-threatening condition.
Introduction
Congenital adrenal hypoplasia (AHC) is a genetic disorder of either autosomal recessive (OMIM#240 200) or an X-linked inheritance (OMIM #300 200). In both inheritance patterns, primary adrenal failure occurs due to defective development of the adrenal gland. In X-linked type, mutations result in permanent lack of adult cortical zone with compensatory changes in residual foetal cells[1] leading to cytomegalic adrenocortical hypoplasia compared to autosomal recessive or sporadic types where there is lack or paucity of both foetal and permanent cortex. Hypogonado- tropic hypogonadism and primary adrenal failure are the key features of the X-linked type AHC due to defective development and functioning of the hypothalamic-pituitary-gonadal and adrenal axis. The presentation is variable; about 60% present with adrenal failure by the third week of life and 40% of them later in childhood. Very rarely those with partial hypogonadism and delayed onset adrenal failure present in adulthood.
The incidence of X-linked AHC is not clearly known. The estimated occurrence may lie between 1:140 000 and 1:1 200 000 children.[2] The NR0B1 gene previously known as DAX1 is mapped on Xp21.2. This gene has two exons with one intervening intron (OMIM database). NR0B1 is an important orphan nuclear receptor involved in normal development and functioning of the hypothalamic-pituitary-gonadal and adrenal axis[3],[4] The product of this gene is a transcriptional factor with the unique feature of lacking the zinc finger DNA binding domain.[5] The gene product plays an important role in development and functioning of the target tissues, namely adrenals and testes. Mutations in the NR0B1 either results in no protein formation or may result in truncated protein when translated. Large deletions have also been reported.[6],[7]
The Cases
Patient 1
The proband came for genetic evaluation at 20 years of age. He was the eldest of three siblings and was well until 3 years of age when he presented with chronic vomiting, skin hyperpigmentation, and failure to thrive. He was hospitalized for the same and found to have hyponatraemia 120 mmol/L (135-145 mmol/L), hyperkalaemia 7.5 mmol/L (3.5-4.5 mmol/L) and hyper- reninaemia 79.4 ng/ml/hour (up to 4.0 ng/ml/hour). A diagnosis of primary adrenal insufficiency was made, and he was started on hydrocortisone and fludrocortisone supplementation with marked improvement. The course was complicated by an Addisonian crisis at 10 years of age. At 15 years of age, he failed to develop secondary sexual characters. The gonadotropin levels were: follicle-stimulating hormone (FSH) 0.915 mIU/ml (0.5-3.3 mIU/ ml); low luteinizing hormone (LH) 0.137 mIU/ml (0.5-3.3 mIU/ ml) and testosterone (<20.0 ng/dl) but normal 17-OH progesterone <0.1 ng/ml (0.0-2.0 ng/ml) which suggest hypogonadotropic hypogonadism with a clinical diagnosis of AHC [Table - 1]. Compliance with replacement was poor as evident by high adrenocorticotropic hormone (ACTH) 952 pg/ml (0.0-46 pg/ml) and growth velocity remained poor. At that time, his age was 17 years and 4 months, and height 147 cm with standard deviation score (SDS) 3.5. The corresponding bone age was 13 years and 6 months (Greulich-Pyle method; G&P), and SDS for mid-parental height (MPH) was 0.8. Family history was positive as two more siblings were affected [Figure - 1].
![[Table - 1]](#tbl_NatlMedJIndia_2019_32_3_141_278692_t2.jpg){kind=link}
![[Figure - 1]](#fig_NatlMedJIndia_2019_32_3_141_278692_f1.jpg){kind=link}
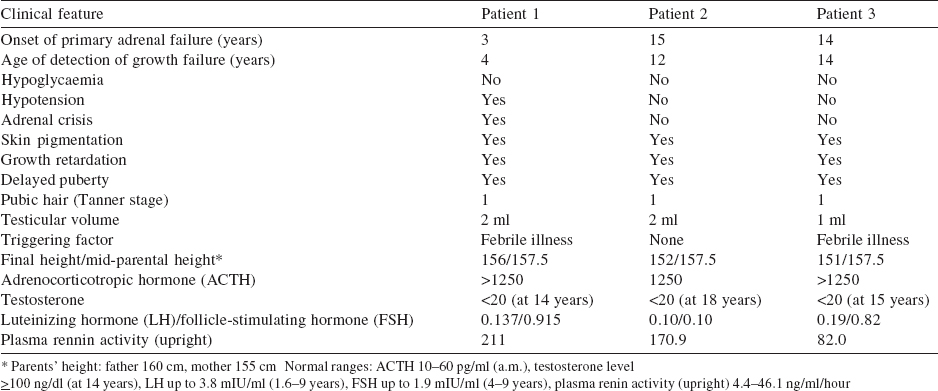
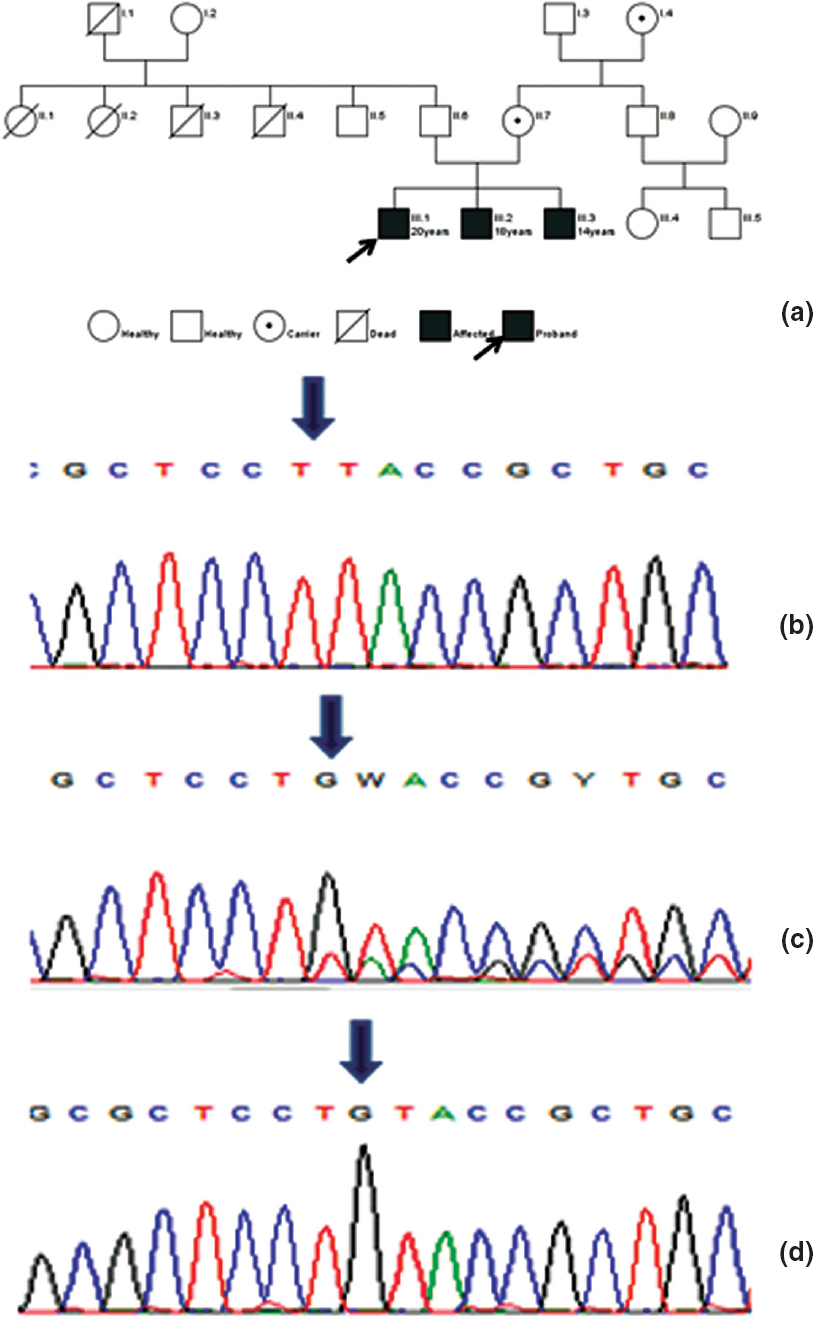 |
| Figure 1: (a) Pedigree; (b) chromatograms showing homozygous frameshift mutation in exon-1 (189_189DelG), arrow placed at deleted G; (c) heterozygous mutation; (d) normal |
Patient 2
The younger brother was 18 years old and first visited the hospital at 5 years with poor appetite, poor weight gain and increased skin pigmentation. His height was 100.5 cm (SDS<1.5). His biochemical profile was normal except for poor response to ACTH stimulation as evident by basal serum cortisol 8.17 μg/dl and 1-hour level of 8.5 μg/dl at the age of 15 years confirming primary adrenal insufficiency. At 18 years, his hormonal profile showed luteinizing hormone (<0.1 mIU/ml), follicle-stimulating hormone (<0.1 mIU/ ml) and testosterone (<20.0 ng/dl) suggesting hypogonadotropic hypogonadism.
Patient 3
The youngest of three siblings was asymptomatic when seen for the first time at our centre at the age of 15 years with a height of 140.5 cm (“3.0 SDS) with a history of upper respiratory tract infection and episodes of vomiting for which he was admitted to a hospital elsewhere. Physical examination showed hyperpig- mented lips, perioral region and bilateral pinna and features of delayed puberty with a testicular volume of 1 ml. Laboratory evaluation showed hyponatraemia, hypocortisolaemia and low gonadotropins indicating primary adrenal failure with hypogonadotropic hypogonadism. He was managed with hydrocortisone and fludrocortisone.
Methods
As the clinical diagnosis of AHC was evident in all three, diagnostic NR0B1 gene analysis was initiated after approval of the institutional review board and taking informed consent from the patients. The genomic DNA was isolated from peripheral blood using Qiagen DNA blood kit. Amplification of exon-1 and exon- 2 including flanking intronic region of NR0B1 was done using a specific set of designed primers. Primers used were as follows: Exon1 AF-AATAGGTCCCAGGAGGCAGC; exon1AR; AAAGCAGCAGCGGTACAGGAGTG; Exon1BF: GACGCTGGGTCCGTGCTG; exon1BR: GACTTCCACAGTCTCGAACTGC; Exon1CF-AGTACTTGCCCTGCTTCCAG; exon1CR-TTCACCTTTGCCCCGACACT; Exon2F-AAGCTAGCAAAGGACTCTGTG; exon2R-TATTTTACACTCTTTTGCCCA. Bi-directional sequencing was carried out on an ABI3500 sequencer. Sequences were compared with the normal reference sequence. Results of molecular testing were interpreted with the patient’s clinical and biochemical profile.
Results
Deletion of G at position 189 (c.189_189delG) resulting in frameshift mutation (Y64Tfs*21) in exon1 of the DAX1 gene leading to premature termination of codon at 84 amino acid position was identified. All three brothers are hemizygous resulting in a truncated protein with 83 amino acids (Fig. 1b-d). Mother and maternal grandmother were heterozygous for the same mutation. The asymptomatic maternal uncle showed wild-type allele. Bioinformatics tools; Mutation taster and Provean were used to predict the nature of the mutation and its consequences. Both bioinformatics tools predicted the mutation as deleterious, the best possible protein structure model showed an alignment score of 63%, but there is existence of nonsense-mediated decay pathways, hence it is difficult to predict the structure and functionality of a mutated protein. Thus, no forward analysis was carried out.
Discussion
AHC is a genetic disorder inherited either as X-linked recessive or as an autosomal recessive pattern. In both inheritance patterns, primary adrenal failure occurs due to defective development of the adrenal gland. The other genetic disorders causing adrenal failure are congenital adrenal hyperplasia with an elevation of 17-OH progesterone, inherited in autosomal recessive manner, X-linked adrenoleukodystrophy associated with white matter disease, behaviour problem and a decline in cognition, and finally AHC with an anatomically small adult cortex of the adrenal gland.
More than 248 (HGMD database) mutations in the DAX1 gene have been reported in the literature, either one amino change, duplication, partial or complete gene deletion[8] rendering the protein non-functional resulting in hypogonadotropic hypo- gonadism.
Intrafamilial heterogeneity was observed in all the three brothers. Primary adrenal failure was observed at different time periods requiring hydrocortisone and fludrocortisone supplementation. However, impaired development of secondary sexual characters due to hypogonadotropic hypogonadism was observed at puberty in all three brothers requiring testosterone replacement therapy. In a previous study, no genotype-phenotype correlation for the hypogonadism was observed.[9]
This novel frameshift mutation does not exist in any database and is reported for the first time in the literature. This mutation analysis provides confirmation of diagnosis and this information can be used for early detection of the disorder and genetic counselling in the family. A larger study for identification of mutations across suspected cases of hypogonadotropic hypogonadism with severe or undiagnosed adrenal insufficiency will help in early detection and management of this life-threatening condition.
Acknowledgement
Department fund 22M030 was used for conducting the tests.
Conflicts of interest. None declared
| 1. | Hay ID, Smail PJ, Forsyth CC. Familial cytomegalic adrenocortical hypoplasia: An X- linked syndrome of pubertal failure. Arch Dis Child 1981;56:715-21. [Google Scholar] |
| 2. | Lin L, Gu WX, Ozisik G, To WS, Owen CJ, Jameson JL, et al. Analysis of DAX1 (NR0B1) and steroidogenic factor-1 (NR5A1) in children and adults with primary adrenal failure: Ten years’ experience. J Clin Endocrinol Metab 2006;91:3048-54. [Google Scholar] |
| 3. | Guo W, Burris TP, McCabe ER. Expression of DAX-1, the gene responsible for X- linked adrenal hypoplasia congenita and hypogonadotropic hypogonadism, in the hypothalamic-pituitary-adrenal/gonadal axis. Biochem Mol Med 1995;56:8-13. [Google Scholar] |
| 4. | Mantovani G, De Menis E, Borretta G, Radetti G, Bondioni S, Spada A, et al. DAX1 and X-linked adrenal hypoplasia congenita: Clinical and molecular analysis in five patients. Eur J Endocrinol 2006;154:685-9. [Google Scholar] |
| 5. | Reutens AT, Achermann JC, Ito M, Ito M, Gu WX, Habiby RL, et al. Clinical and functional effects of mutations in the DAX-1 gene in patients with adrenal hypoplasia congenita. J Clin Endocrinol Metab 1999;84:504-11. [Google Scholar] |
| 6. | Rojek A, Krawczynski MR, Jamsheer A, Sowinska-Seidler A, Iwaniszewska B, Malunowicz E, et al. X-linked adrenal hypoplasia congenita in a boy due to a novel deletion of the entire NR0B1 (DAX1) and MAGEB1-4 genes. Int J Endocrinol 2016;2016:5178953. [Google Scholar] |
| 7. | Khadilkar VV, Mangtani HR, Jahagirdar RR, Khatod KA, Phadke ND, Deepa PS, et al. Entire DAX1 gene deletion in an Indian boy with adrenal hypoplasia congenita. Indian J Pediatr 2013;80:631-5. [Google Scholar] |
| 8. | Rabiee M, Jalali E. Infantile-onset acute primary adrenal insufficiency: X linked adrenal hypoplasia congenital: A molecular analysis of DAX1. IIOABJ 2016;7: 105-11. [Google Scholar] |
| 9. | Rojek A, Obara-Moszynska M, Malecka E, Slomko-Jozwiak M, Niedziela M. NR0B1 (DAX1) mutations in patients affected by congenital adrenal hypoplasia with growth hormone deficiency as a new finding. J Appl Genet 2013;54:225-30. [Google Scholar] |
Fulltext Views
1,100
PDF downloads
310



