
Translate this page into:
Primer of Epidemiology IV. Study designs II: Interventional or experimental designs
To cite: Singh K, Gupta P, Shivashankar R. Primer of Epidemiology IV. Study designs II: Interventional or experimental designs. Natl Med J India 2021;34:228–31.
Abstract
In this article, we describe experimental study designs and focus on randomized controlled trials. Experimental studies are intervention studies in which the investigator tests a new treatment on a selected group of patients. In a controlled design, the effects of an intervention (new treatment) are measured by comparing the outcome in the experimental group with that in a control group. Experimental studies are similar to cohort studies except that the exposure is a deliberate change (intervention) made by the researcher in one group of participants and it overcomes confounding because the treatment is assigned randomly. Further, we discuss various types of randomization (random sequence allocation) and importance of allocation concealment and blinding for proper assessment of outcomes in randomized trials.
JAMES LIND SCURVY STUDY: A HISTORICAL RANDOMIZED TRIAL
James Lind was a Scottish naval surgeon. During the early 18th century, thousands of people were dying of scurvy and it was a major cause of death among sailors serving the Royal Navy. In 1747, on board HMS Salisbury, he carried out experiments to discover the cause of scurvy, the symptoms of which included loose teeth, bleeding gums and haemorrhages. It is considered one of the first controlled clinical trials recorded in medical science. He selected 12 sailors suffering from similar symptoms of scurvy, divided them into 6 pairs and treated them with previously suggested remedies. The first five pairs of sailors received a quart of cider daily; 25 gutts of elixir vitriol three times a day; two spoonsful of vinegar three times a day; half a pint of seawater daily and a concoction of nutmeg, mustard and garlic three times a day. The sixth pair was prescribed two oranges and a lemon daily. He reported that by the end of the week, those on citrus fruits recovered well, while the others did not show any improvement. Although there is no information on how treatment allocation was done among the pair of sailors, he took care of selection bias, noting that potential confounding factors— clinical condition, basic diet and environment—had been held constant.1,2
ANTURANE REINFARCTION TRIAL: SULPHINPYRAZONE IN THE PREVENTION OF SUDDEN DEATH AFTER MYOCARDIAL INFARCTION
How do withdrawals affect reported findings?
The Anturane Reinfarction Trial (ART) is an example of how withdrawal of randomized study participants in a clinical trial can favourably influence its reported results. The US Food and Drug Administration took the unusual step of criticizing the sponsor in an article published in the New England Journal of Medicine. The ART aimed to determine whether the platelet-active drug sulphinpyrazone (anturane) improved prognosis over a 2-year period among survivors of acute myocardial infarction (MI). One of the major criticisms of the trial was on the withdrawal of 71 of 1629 randomized study participants from the final data analysis. The authors claimed that these 71 participants did not meet the eligibility criteria of the study. Of the 71 withdrawals, 38 had been randomized to the sulphinpyrazone group and 33 had been assigned to the placebo group––hardly a difference that would warrant attention. However, 10 of the 38 (26.3%) withdrawn patients on sulphinpyrazone died versus only 4 of the 33 (12.1%) withdrawn patients on placebo. The difference in the number of deaths among study participants withdrawn from the analysis contributed to the reported statistically significant mortality results favouring sulphinpyra-zone. Following the controversy, intention-to-treat analysis has become mandatory in all clinical trials.3,4
Experimental studies are intervention studies in which the investigator tests a new treatment on a selected group of patients. In a controlled design, the effects of an intervention (new treatment) are measured by comparing the outcome in the experimental group with that in a control group. Experimental studies are similar to cohort studies except that the exposure is a deliberate change (intervention) made by the researcher in one group of participants. Importantly, experimental designs overcome the errors due to confounding because the treatment is assigned randomly.
A randomized controlled trial (RCT), in which patients are randomly allocated (see the section on ‘Randomization’) to the intervention or control group, is among the most rigorous study designs in epidemiology. Through the randomization process, study participants in the intervention and treatment groups are expected to be comparable on potential confounding factors that might otherwise bias observed associations (Fig. 1). The effect of the intervention is quantified by comparing outcomes between the two treatment groups. For example, use of multidrug pill in reducing cardiovascular events trial was an open-label, randomized, controlled, multicentre trial involving 2004 participants (including 1000 in India and 1004 in Europe [the United Kingdom, the Netherlands and Ireland]) with established cardiovascular disease or those at high risk of cardiovascular disease.5 Eligible patients were either randomized to receive the polypill-based treatment strategy or continued with their usual care and were followed up for at least 1 year. The primary outcomes of the study were to assess the effect of polypill strategy on adherence to indicated medications (defined as self-reported current use of antiplatelet, statin and combination (≥2) blood pressure [BP]-lowering therapy), change in BP and change in low-density lipoprotein cholesterol (LDLc), from baseline to end-of-trial follow-up.4
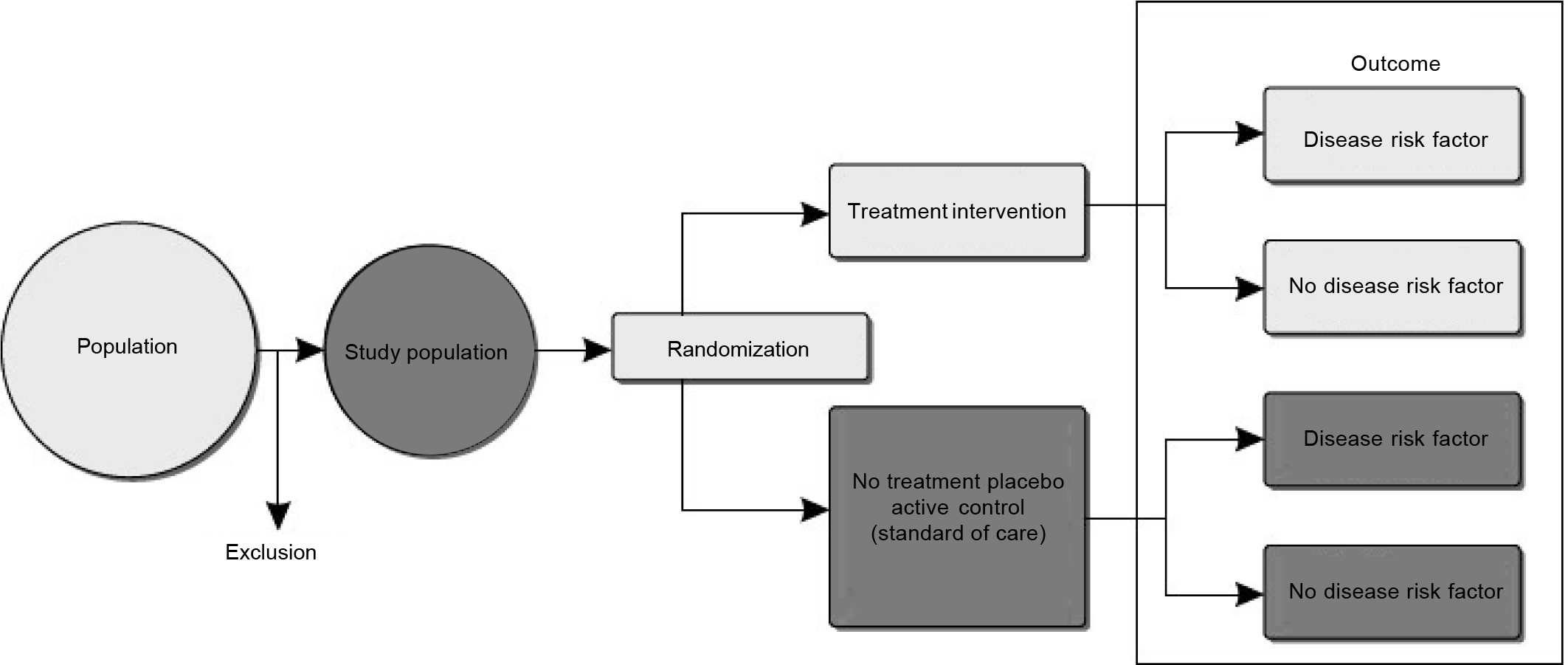
- Design of experimental or intervention study
Another example of an individual RCT is the National Heart Lung Blood Institute-funded CARRS trial, which randomized 1146 poorly controlled patients with type 2 diabetes mellitus attending 10 diabetes clinics in India and Pakistan to a multicomponent quality improvement intervention (consisting of non-physician care coordinators to enhance patients’ adherence and clinical decision-support software to improve physician’s responsiveness) versus usual care for cardiovascular risk reduction.6 The primary outcome of the study was to assess the proportion of intervention versus usual care group participants achieving multiple risk factor control, i.e. glycated haemoglobin (HbA1c <7%) and BP (<130/80 mmHg) and/or LDLc <100 mg/dl. Field trials evaluate the interventions aimed at reducing exposure (risk factor) without necessarily measuring the health effects. Thus, it involves people who are healthy but presumed to be at risk and data collection takes place ‘in the field’. (For example, the D-CLIP trial assessed the effect of lifestyle interventions compared with that of metformin or no treatment in population at risk of incident diabetes in Chennai, India.)7
In a community trial, a community rather than an individual is randomized to the treatment or control groups. This is particularly useful when the intervention under study is delivered at the community level and it is challenging to ensure that individuals will adhere to the assigned treatment group. For example, the DISHA study is evaluating the effect of task-shifting interventions involving frontline health workers for cardiovascular risk reduction in the community in India.8
Methods of outcome assessment in clinical trials are crucial to reduce the risk of ascertainment of bias in effectiveness of treatment. As a best practice, RCTs evaluating the effect of intervention on hard clinical end-points (e.g. cardiovascular death, fatal or non-fatal MI, stroke, heart failure, renal failure, diabetic retinopathy) will constitute an end-point adjudication committee to review the supporting documentation provided by the treating physicians. The role of a blinded end-point adjudication committee is to independently review the patient’s medical reports (e.g. hospital admission and discharge summary, blood reports, electrocardiogram) and classify the end-point as a true end-point or false if the reported diagnosis matches with the standard definition given as per the international guidelines. This avoids the potential of incorrect diagnosis and subjective errors in classifying an end-point such as MI due to difference in local practices. In resource-constrained studies where hard clinical end-points cannot be studied in a large clinical trial, intermediate outcomes such as changes in BP, glycaemia and lipids can be studied. Associated risk of bias with assessment of intermediate outcomes does not necessitate adjudication as these are mostly objective measures (but important to ensure that the laboratories meet minimum standards of quality assurance) and are verified by medical/laboratory reports during random on-site monitoring visits. However, when subjective measures of the outcome, e.g. change in quality-of-life score, depression score or pain score, are considered as the primary outcomes of the study, it is recommended that outcomes are assessed by an independent (blinded to treatment assignment) outcome assessor not directly involved in the study to reduce the risk of bias in measurement of outcomes.
The primary advantage of an RCT is that proper randomization will on average eliminate potential bias in the study by equally distributing the confounding factors between the intervention and control groups, such that groups are comparable at the start and any differences observed in other variables at the end of trial is due to chance (unaffected by conscious/unconscious biases of investigators).
Some limitations of randomized trials are volunteer bias: the study population is not a true representative of the target population, which implies generalizability issues of study findings. Furthermore, there are ethical and practical concerns, for example exposing patients to an inferior or harmful intervention than the current treatment is often thought unethical. Limitations of community trials are that it is difficult to isolate the communities where the intervention is being delivered from general social changes that may be taking place simultaneously, and favourable changes in risk factors in control sites, which may attenuate the true effects of the intervention. Furthermore, often a small number of communities/ clusters are available and random allocation is not feasible.
Randomization
In experimental research, randomization is the procedure used to allocate individuals to a particular intervention. The term randomization is different from random sampling. In random sampling, there is equal probability for a participant to be selected for the study. In randomization, there is equal probability for participants to be assigned to a particular intervention. In simplistic terms, which intervention a participant will receive is decided by a toss of coin. This process aims to reduce selection bias because participants, recruiters, physicians or researchers will not determine which group the participant will be assigned. Furthermore, if randomization is done properly, in a sufficient sample size, it is expected that confounding factors (both known and unknown) will be distributed equally between the intervention and control groups. Therefore, the observed differences in outcomes beyond chance can be directly attributed to the intervention.
Randomization involves two important processes: (i) generation of random sequence for allocation and (ii) allocation concealment or blinding.
Random sequence for allocation
This is the process of allocation of intervention to a sequence of participants. The following are the common methods:
Simple random allocation. This is the simplest form of random allocation wherein each participant is allocated to intervention or control groups at a toss of coin or an equivalent method (in present days, most experimental trials use computer-based random allocation). This straightforward method produces a completely unpredictable sequence. The major disadvantage of this method is that this may result in unequal distribution of participants in intervention groups specifically if the sample size is small. This would result in reduced statistical power and render the study fruitless. For example, in a study of 30 patients, by sheer chance, more than 20 patients may end up in the control group, causing imbalance between the number of individuals in the intervention and control groups.
Blocked random allocation. This is also known as restricted random allocation, which is designed to ensure that the number of participants is equally distributed between the intervention and control groups. Here, participants are randomized within blocks of 4, 6 or 8, ensuring equal distribution within each block. In the blocked random method, in addition to ensuring equal distribution of participants between the groups, there are other advantages. If the types of participants vary over the recruitment period of the study (e.g. more severe form of cases usually occur in winter), this will ensure equal distribution between groups. Furthermore, if the study had to be stopped prematurely, this method will still provide balanced numbers between groups at any point in the study. One major disadvantage of blocked random allocation is that in trials where physicians are not blinded to allocation, the block size and intervention assignment for the last participant in each block are predictable. For example, in a two-group trial, if the block size is 6, and the first five assignments are group 1, group 1, group 2, group 2 and group 2, the last assignment will be 100% predictable, i.e. group 1. Therefore, in the studies that are not double-blinded (i.e. both the investigator and patients are aware of the treatment strategy), random allocation can be done using blocks of variable size. That is, size of blocks is randomly varied during allocation, rendering it difficult to predict allocation of intervention.
Stratified random allocation. In studies that have a small number of participants, it may be important to ensure that the subgroups of participants are equally distributed (e.g. age groups and gender). In these cases, random allocation can be done by stratification, ensuring a balanced number of participants with characteristics in each group. This can also be done in multisite studies to achieve balance in allocation groups for all the sites.
Another variant of stratified random allocation is matched pair randomization, specifically used for cluster randomized trials. In this method, a cluster such as a village is matched with another cluster for predefined variables (population, percentage of families working in agriculture, distance of major road, etc.). After matching, interventions are allocated randomly.
In all the types of random allocation, it is crucial to ensure that the recruiting physician does not know the allocation list. The allocation list should be prepared centrally by a person who is not involved directly in the study (e.g. independent trial statistician or external agent). The allocation can be done over the telephone or using applications on computers.
Allocation concealment or blinding
If the participants or physicians are aware of the allocation, then there is a possibility of observational bias. Whenever practically feasible, it is important to conceal the allocation of intervention to both participants and physicians. This is called double blinding. However, in some cases, physicians cannot be blinded to interventions (e.g. trials that involve two different approaches to a surgical procedure). In such cases, only patients are blinded to allocation. This is called single blinding. Sometimes, the analysts are blinded to allocations until the final analysis to prevent personal bias during analysis. When all three–– participants, physician and analysts––are blinded to allocation, it is called triple blinding.
ACKNOWLEDGEMENTS
We are grateful to Dr D. Prabhakaran, Vice President for Research, Public Health Foundation of India (PHFI) for critical inputs in developing this manuscript. We also thank Ms Sanjana Bhaskar, Research Assistant, Centre for Environment Health, PHFI, for editorial assistance and referencing.
Conflicts of interest
None declared
References
- James Lind's treatise of the scurvy 1753. Postgrad Med J. 2002;78:695-6.
- [CrossRef] [PubMed] [Google Scholar]
- Documenting the evidence: The case of scurvy. Bull World Health Organ. 2004;82:791-6.
- [Google Scholar]
- Sulfinpyrazone in the prevention of sudden death after myocardial infarction. N Engl J Med. 1980;302:250-6.
- [CrossRef] [PubMed] [Google Scholar]
- The FDA's critique of the anturane reinfarction trial. N Engl J Med. 1980;303:1488-92.
- [CrossRef] [PubMed] [Google Scholar]
- Effects of a fixed-dose combination strategy on adherence and risk factors in patients with or at high risk of CVD: The UMPIRE randomized clinical trial. JAMA. 2013;310:918-29.
- [CrossRef] [PubMed] [Google Scholar]
- Effectiveness of a multicomponent quality improvement strategy to improve achievement of diabetes care goals: A randomized, controlled trial. Ann Intern Med. 2016;165:399-408.
- [CrossRef] [PubMed] [Google Scholar]
- The stepwise approach to diabetes prevention: Results from the D-CLIP randomized controlled trial. Diabetes Care. 2016;39:1760-7.
- [CrossRef] [PubMed] [Google Scholar]
- Task shifting of frontline community health workers for cardiovascular risk reduction: Design and rationale of a cluster randomised controlled trial (DISHA study) in India. BMC Public Health. 2016;16:264.
- [CrossRef] [PubMed] [Google Scholar]



