Translate this page into:
A 45-year-old woman with episodic anxiety, hypertension and diabetes mellitus
Corresponding Author:
| How to cite this article: . A 45-year-old woman with episodic anxiety, hypertension and diabetes mellitus. Natl Med J India 2018;31:160-163 |
The Case
A 45-year-old woman was admitted at another hospital with complaints of episodic anxiety and sweating for 2 years, hypertension (HTN) and diabetes mellitus (DM) for 2 months, hirsutism for 2 months, and swelling over her hands and feet, difficulty in walking, getting up from bed and climbing stairs for 20 days.
She had no history of hyperpigmentation or abdominal striae. There was no history of neurological or muscular weakness. Her past and family histories were non-contributory. The records provided by the admitting hospital showed that, at the time of admission, she was sick and had HTN (blood pressure [BP] 190/ 114 mmHg). There was no hyperpigmentation or abdominal striae on examination; however, hirsutism was present.
She had hypokalaemia (K+ 2.9 mEq/L), hyperglycaemia (random blood sugar 373 mg/dl) and metabolic alkalosis. She had leucocytosis, but there was no fever. Chest X-ray was normal. Blood culture grew Staphylococcus hominis while urine culture showed Enterococcus sp. Further investigations revealed secondary hypothyroidism (serum T4 2.10 μg/dl and thyroid-stimulating hormone 0.15 μIU/ml). A basal 8 a.m. serum cortisol was 107 μg/ dl with increased plasma adrenocorticotropic hormone (ACTH) level (538.9 pg/ml; normal 9-55 pg/ml). Her overnight dexa- methasone suppression test did not show any suppression of basal cortisol [Table - 1].
![[Table - 1]](#tbl_NatlMedJIndia_2018_31_3_160_255755_t7.jpg){kind=link}

Contrast-enhanced computed tomography (CECT) scan of the abdomen showed bilateral bulky adrenal glands and a well- defined hypodense lesion measuring 3 cmx2 cmx2 cm with a surrounding enhancing rim involving the left adrenal gland [Figure - 1] and [Figure - 2]. Magnetic resonance imaging (MRI) of the brain showed a partial empty sella [Figure - 3], and the CECT chest was normal.
![[Figure - 1]](#fig_NatlMedJIndia_2018_31_3_160_255755_f1.jpg){kind=link}
![[Figure - 2]](#fig_NatlMedJIndia_2018_31_3_160_255755_f2.jpg){kind=link}
![[Figure - 3]](#fig_NatlMedJIndia_2018_31_3_160_255755_f3.jpg){kind=link}
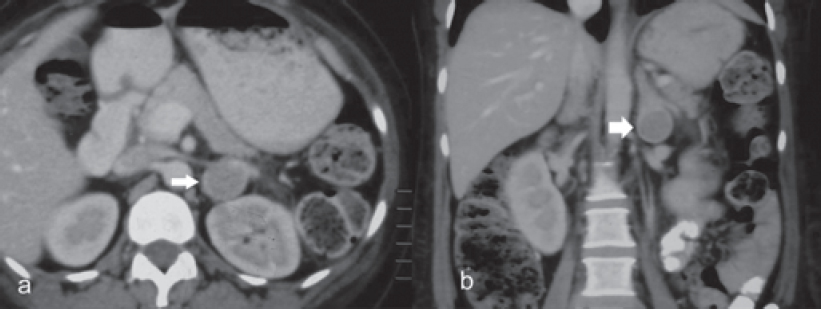 |
| Figure 1: CT images showing left adrenal nodule (a) axial, (b) coronal |
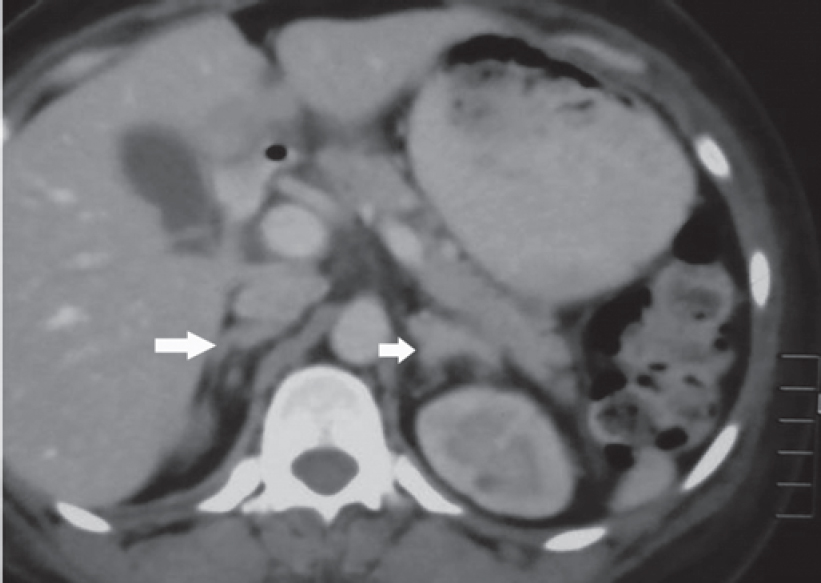 |
| Figure 2: CT images showing bulky bilateral adrenal glands |
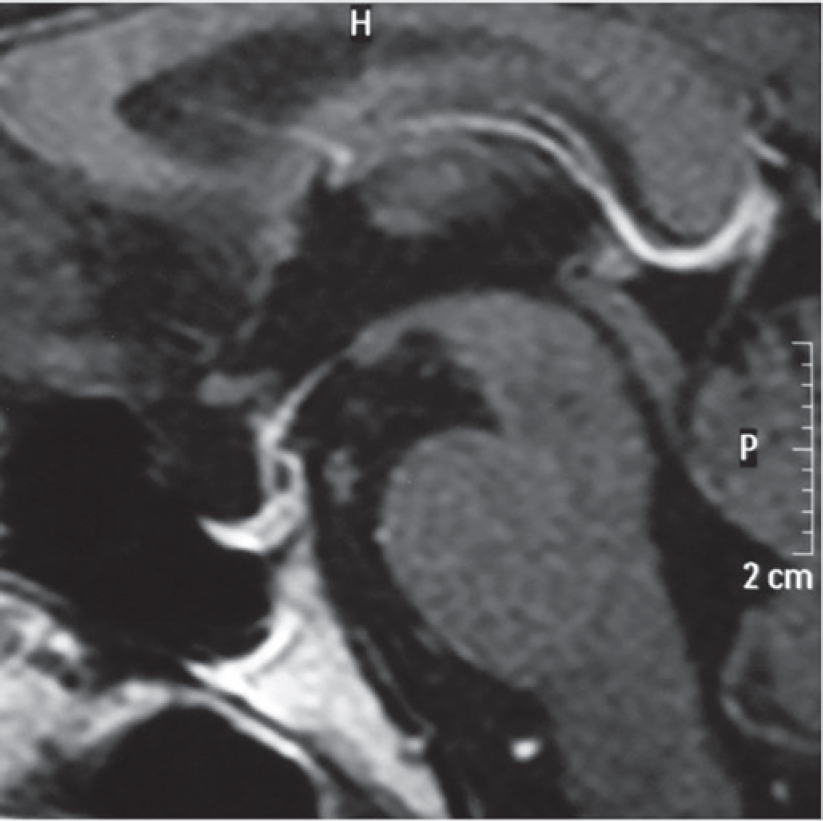 |
| Figure 3: Sagittal magnetic resonance imaging of the brain showing normal pituitary |
She was managed conservatively with antibiotics, antihypertensive medications, insulin and levothyroxine. After symptomatic improvement, she was brought to our emergency room with complaints of breathlessness, restlessness and increased thirst. She was conscious, oriented and her pulse was 100/minute, BP was 160/110 mmHg and respiratory rate was 24/minute. Her oxygen saturation was 97% and the chest was clear on auscultation.
She continued to have leucocytosis and thrombocytopenia and was started on intravenous antibiotics, insulin, antihypertensives and was taken up for surgery after optimization.
Differential Diagnosis
Dr Gaurav Agarwal: This patient presented with recent-onset HTN without any family history. When we add to it the recently diagnosed DM, this indicates some metabolic or endocrine cause. When we also consider the symptoms of hirsutism, proximal muscle weakness and water retention, a few differential diagnoses can be considered. It could be type 2 DM with HTN, or polycystic ovary syndrome or she could well be just obese. However, Cushing syndrome should be considered in this clinical picture even though there is no hyperpigmentation or striae.
The patient had episodic sweating and anxiety. In conjunction with HTN, this raises the possibility of a pheochromocytoma, although these symptoms could be explained by DM. The investigations provided by the first hospital show hypokalaemia, metabolic alkalosis and hyperglycaemia. Thyroid function tests showed features of secondary hypothyroidism. Serum cortisol remained unsuppressed on the overnight dexamethasone suppression test, and her ACTH levels were significantly raised, which is indicative of ACTH-dependent hypercortisolism. The source of ACTH needs to be confirmed—whether it is from the pituitary or an ectopic source.
The presence of secondary hypothyroidism with ACTH- dependent Cushing syndrome suggests a pituitary source; however, the absence of hyperpigmentation is a factor against it. The points in favour of ectopic ACTH secretion are high level of ACTH, severe hypercortisolism, hypokalaemia and metabolic alkalosis. To confirm hypercortisolism, we need low- or high-dose dexamethasone suppression test or late-night salivary cortisol. We further need pituitary imaging to rule out pituitary micro- or macro-adenoma. If we do not find a pituitary lesion, then we may perform inferior petrosal sinus sampling (IPSS) for ACTH with corticotrophin-releasing hormone (CRH) stimulation and we need imaging to locate the possible source of ectopic ACTH. This can be in the form of CT of the chest to rule out any lung mass and whole-body Ga68 DOTANOC positron emission tomography (PET)-CT. I would like to review the available imaging.
Dr Sanjay Sharma: We have abdominal imaging in the form of a single-phase CECT of the abdomen and pelvis which shows that both the adrenals are prominent, left more hyperplastic than the right [Figure - 1] and [Figure - 2]. In addition, the left adrenal shows a mass arising from the lateral limb with some peripheral enhancement suggestive of a capsule [Figure - 1]. There is no mass elsewhere in the retroperitoneum and in the area around the aortic bifurcation. CECT chest does not show any mass in relation to the tracheo- bronchial tree, in the mediastinum or elsewhere. The imaging of the brain was done in the form of CT and MRI of the brain, which was unremarkable [Figure - 3]. The imaging of the sella does not show any features of a pituitary adenoma.
Dr Gaurav Agarwal : Thank you Dr Sharma. In a nutshell, we have a patient with recent-onset diabetes and HTN, ACTH- dependent Cushing syndrome along with bilateral adrenal hyperplasia and a left adrenal mass. MRI brain is normal, so we are not dealing with a patient who has Cushing disease. The next logical step is to look for the source of ectopic ACTH. We could now review the IPSS or DOTANOC PET-CT if these were done.
Dr Rajeev Kumar: We did not do IPSS, but Ga68 DOTANOC PET-CT showed intense uptake in the left adrenal lesion and no uptake anywhere else in the body.
Dr Gaurav Agarwal: This was an important finding. A high concentration of Ga68 DOTANOC avidity of the adrenal lesion is highly suggestive of a neuroendocrine tumour, consistent with the diagnosis of pheochromocytoma.
That was the stage when the patient pre sented to the emergency department of the All India Institute of Medical Sciences (AIIMS) with complaints of breathlessness, restlessness and increased thirst. She was hypertensive and looking sick. Investigations revealed hypokalaemia and metabolic acidosis. She had leucocytosis and low platelets which might be indicative of an ongoing infective process as her previous urine and blood cultures were positive. The high serum ACTH levels and hypercortisolism were confirmed here. However, the most interesting new finding is the high level of urinary metanephrine (1415.7 μg/ml).
We now have an added dimension in a patient with ectopic ACTH-dependent Cushing syndrome—high levels of metanephrine! The high level of metanephrine is not just a marginal rise that could be attributed to a false-positive report. It is almost four times the upper limit of the normal range and in all likelihood suggests a catecholamine-producing tumour. Hence, based on the clinical details, investigations and imaging, the possibilities are ACTH-dependent Cushing syndrome, or pheochromocytoma, or both.
Two scenarios are possible. First, there may be two separate pathologies, i.e. ACTH-dependent Cushing syndrome coexisting with a pheochromocytoma. This may be either in the form of a pituitary adenoma with pheochromocytoma or an ectopic ACTH- producing tumour with pheochromocytoma. However, there is no case report in the English literature till date about either of these scenarios. This may be due to rarity of both of these conditions individually, which makes it very rare for both lesions to coexist. I believe it is only a matter of time that such a case is reported.
The second scenario for the coexistence of both of these conditions can be an ectopic ACTH-secreting pheochromocytoma or a collision adrenal tumour. Even though it looks a bit unusual, such cases have been reported in the literature. ACTH-secreting pheochromocytomas are known to occur for more than a few decades now with around two dozen published case reports. A similar case report published in March 2016 from Japan[1] reported a patient with a left pheochromocytoma (5 cm) with bilateral adrenal hyperplasia. The most interesting aspect was the ACTH staining in the same sections showing staining typical for neuroendocrine tumours such as synaptophysin. A similar case has been reported from India, where pheochromocytoma was associated with subclinical Cushing syndrome.[2] We too have managed a similar patient at the Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow, and probably due to that experience, I can suggest a possible diagnosis. There has also been a case report of Cushing syndrome caused by secretion of precursors of ACTH by a pheochromocytoma, rather than ACTH itself.[3]
The other possible scenarios may be an ectopic ACTH-secreting tumour with a non-functional adrenal adenoma with falsely elevated metanephrine levels or a collision adrenal carcinoma causing both ACTH and catecholamine secretion. It could also be a pseudo-pheochromocytoma with Cushing syndrome but does not seem to be likely in this scenario.
Hence, with the clinical scenario and the investigation results provided, my diagnosis for this patient will be an ACTH-secreting pheochromocytoma. This is a rare condition with about two dozen case reports published in the literature, but I also think that this condition is under-reported because we usually do not obtain serum cortisol or ACTH levels in patients with pheochromo- cytomas. In addition, ACTH staining is not done in every patient with a pheochromocytoma because of the cost and its availability at select centres.
Dr GAURAV AGARWAL ' S DIAGNOSIS
– ACTH-secreting pheochromocytoma
Clinical Discussion
Dr A.K. Dinda: Thank you Dr Agarwal for such a thorough discussion. Before revealing the final diagnosis, I would like to invite any questions from the house to our clinical discussor.
Audience : Can the raised metanephrine levels be due to stress- induced physiological catecholamine secretion?
Dr Gaurav Agarwal: Yes, I did consider the possibility. Severe hypercortisolism is a stressful condition and can trigger physiological catecholamine secretion. However, looking at the literature, in such conditions the metanephrine levels are usually elevated less than two times the upper limit of normal. In our patient, metanephrine levels were raised almost four times the upper limit of normal. Such high levels are unlikely to be due to physiological catecholamine secretion.
Pathological Discussion
Dr A.K. Dinda: We received bilateral adrenalectomy specimens. On gross examination, the left adrenal showed a nodule in the lower pole measuring 3 cmx2.5 cmx2 cm with the rest of the adrenal gland enlarged suggestive of adrenal hyperplasia [Figure - 4]. The right adrenal gland also was diffusely hyperplastic [Figure - 4]. Upon microscopic examination of the left adrenal cortex [Figure - 5], there were homogeneous areas of cells containing eosinophilic cytoplasm and vesicular nuclei forming nodules at places. There was no cellular atypia or mitoses, and the overall features were of adrenocortical hyperplasia.
![[Figure - 4]](#fig_NatlMedJIndia_2018_31_3_160_255755_f4.jpg){kind=link}
![[Figure - 5]](#fig_NatlMedJIndia_2018_31_3_160_255755_f5.jpg){kind=link}
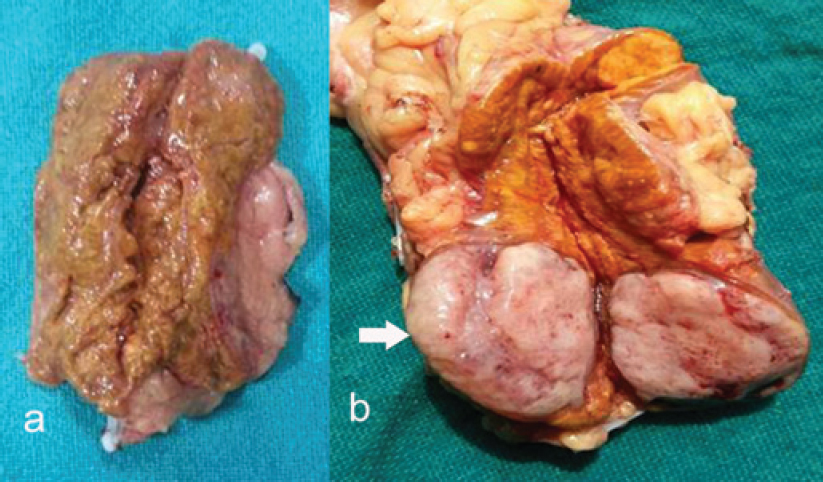 |
| Figure 4: Gross examination of resected adrenals, (a) right enlarged gland, (b) left adrenal nodule |
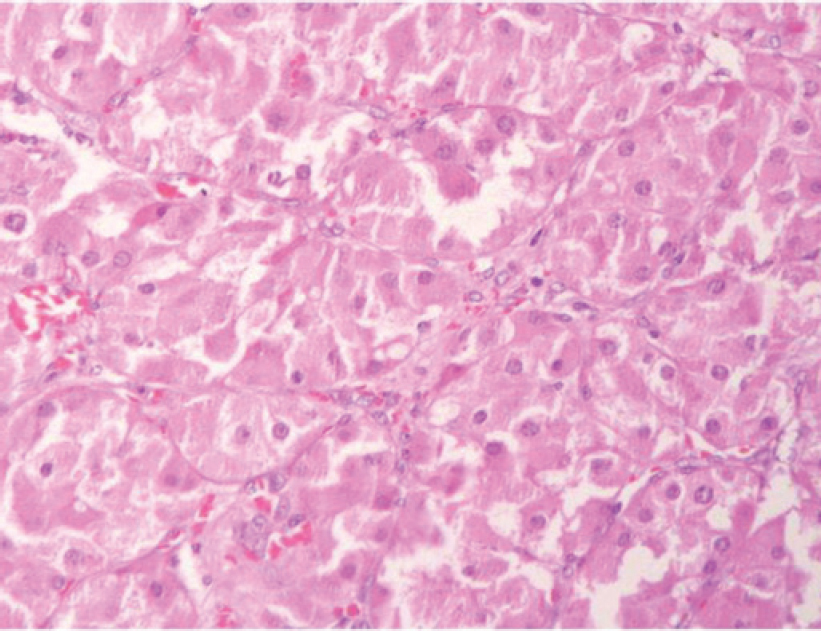 |
| Figure 5: Section from the left adrenal nodule showing cells with eosinophilic granular cytoplasm (oncocytic change), vesicular nucleus and no atypia (H and E) |
The left adrenal nodule showed monomorphic cells with a polygonal shape, granular cytoplasm and vesicular nuclei with no cellular atypia. This appearance is typically seen in a pheochromo- cytoma. We did immunohistochemistry to confirm the medullary origin of this tumour and whether it was secreting other hormones that could be responsible for the clinical features of this patient. The tumour stained positive for synaptophy sin and chromogranin, confirming it to be a pheochromocytoma while it was negative for calretinin and inhibin, ruling out the adrenal cortical origin [Figure - 6]. It also had positive staining for ACTH but was negative for CRH [Figure - 6]. The right adrenal gland showed microscopic features of cortical hyperplasia while the medulla was unremarkable.
![[Figure - 6]](#fig_NatlMedJIndia_2018_31_3_160_255755_f6.jpg){kind=link}
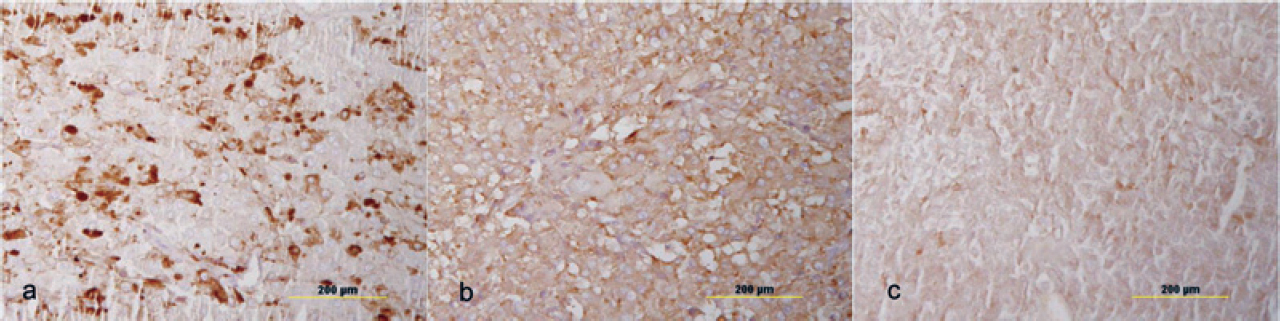 |
| Figure 6 Immunohistochemistry of the left adrenal nodule showing positive staining for (a) chromogranin, (b) synaptophysin and (c) adrenocorticotropic hormone |
Final diagnosis
–ACTH-secreting left pheochromocytoma with cortical hyperplasia of both the adrenals
Concluding Discussion
Dr A.K. Dinda: Adrenocorticotropic hormone-secreting pheochromocytomas are rare tumours. About 32 cases have been reported in the literature.[4] These tumours have a female predilection (70%) and all except four cases were reported in adults. Except for one case, all these tumours were benign. The diagnostic criteria for ACTH-secreting pheochromocytoma were established by Chen et al.[5] in 1995. These include (i) clinical and laboratory evidence of hypercortisolism; (ii) elevated plasma ACTH level; (iii) evidence of a pheochromocytoma by urinary catecholamines, metanephrines and vanillylmandelic acid or MRI evidence of an adrenal mass with a bright T2 signal; (iv) resolution of symptoms and signs of adrenocorticoid and catecholamine excess after unilateral adrenalectomy; and (v) rapid normalization of the plasma ACTH levels after adrenalectomy. Now, I will invite Dr Rajeev Kumar to tell us what happened after the surgery because the last two criteria are important for the confirmation of diagnosis.
Dr Rajeev Kumar: Thank you, Dr Dinda. I must congratulate Dr Agarwal for getting the diagnosis absolutely right. This patient was admitted to the Department of Endocrinology, and we were involved in doing the bilateral adrenalectomy. When we saw the patient, she was extremely unwell with many challenging issues before us. She had thrombocytopenia and leucocytosis that were most likely due to sepsis. She was managed with broad-spectrum intravenous antibiotics. She also required single-donor platelets before we could take her up for surgery. She had severe hypo- kalaemia requiring both intravenous and oral supplementation. She also had severe proximal myopathy. Hence, our assessment was that she was a high-risk candidate for surgery. Our anaesthetist, Dr Rashmi Ramachandran, was involved in managing these issues and optimizing the patient for surgery. We were able to do the bilateral adrenalectomy 3 weeks after admission.
We did a bilateral simultaneous laparoscopic adrenalectomy, first on the left side because it clearly showed a tumour, followed by the right side through a standard transperitoneal approach. There were no intraoperative complications and the blood loss was 200 ml. She required one packed red blood cell and one single-donor platelet transfusion. She was haemodynamically stable throughout the postoperative period and did not require inotropic support. She was kept in the intensive care unit on the day of surgery and shifted to the ward on postoperative day 1.
She was started on normal oral diet on postoperative day 1, but she did require low-dose antihypertensives and insulin for 1 week. She also required potassium supplementation and diuretics for generalized oedema. ACTH levels were obtained on postoperative day 5, which were <0.01 pg/ml. Total leucocyte and platelet counts normalized within the first week without any intervention.
Unfortunately, she developed acute left retinal necrosis on postoperative day 8 and was evaluated; this was considered to be due to metastatic endophthalmitis. She required left vitrectomy and intra vitreal antibiotics and underwent buckling surgery and abscess drainage a few days later, but the vision of the left eye did not return.
At present, 1 year after surgery, she is normotensive and normoglycaemic without any medications. She requires steroid and thyroxine supplementation; however, there has been no improvement in the vision of the left eye.
AUDIENCE : Why did you do bilateral adrenalectomy when there was a nodule in the left adrenal?
Dr Rajeev Kumar: In hindsight, it does seem that left adrenalectomy alone may have been sufficient and we could have assessed the ACTH levels. However, this patient had a stormy preoperative course and she was optimized for surgery with great difficulty. In fact, just 1 week before the surgery, she had an episode of probable cardiac arrest due to arrhythmia secondary to severe hypokalaemia for which she required cardiopulmonary resuscitation for a brief period. We initially intended to perform unilateral adrenalectomy but were concerned that she may not withstand another surgery if the nodule in the left adrenal was not the source of ACTH secretion. Removing both adrenals would ensure resolution, whatever may be the source of ACTH. Therefore, after much discussion among us, we decided to go ahead with bilateral adrenalectomy.
Dr Gaurav Agarwal : Do you use adrenolytic drugs such as ketoconazole or mitotane to control severe hypercortisolism in such patients?
Dr Rajesh Khadgawat: We do use ketoconazole in some patients who have severe Cushing syndrome but not mitotane because it is not easily available. Our experience with ketoconazole shows that approximately half these patients develop liver dysfunction and we are forced to stop it. However, this patient was too sick to be started on ketoconazole and the quickest way to control hypercortisolism was bilateral adrenalectomy.
ACTH-producing pheochromocytomas are extremely rare tumours, but the presence of hypercortisolism with elevated ACTH and typical features, clinical and biochemical of pheochromocytoma, should bring this diagnosis into consideration. A high-risk surgery can be life-saving if sufficient team effort and infrastructure are available to manage the patient.
Acknowledgement
We thank Dr Rashmi Ramachandran, Department of Anaesthesiology and Critical Care, All India Institute of Medical Sciences, New Delhi, for her support.
Conflicts of interest. None declared
Compiled by PRADEEP PRAKASH, RAJEEV KUMAR, AMIT K. DINDA
Participants
All India Institute of Medical Sciences, Ansari Nagar, New Delhi 110029, India
Pradeep Prakash, Rajeev Kumar Department of Urology Rajesh Khadgawat Department ofEndocrinology and Metabolism
Amit K. Dinda Department of Pathology
Sanjay Sharma Department of Radiodiagnosis
Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow, Uttar Pradesh, India
Gaurav Agarwal Department of Endocrine and Breast Surgery
Correspondence to Rajeev Kumar; rajeev02@gmail.com
| 1. | Sakuma I, Higuchi S, Fujimoto M, Takiguchi T, Nakayama A, Tamura A, et al. Cushing syndrome due to ACTH-secreting pheochromocytoma, aggravated by glucocorticoid- driven positive-feedback loop. J Clin EndocrinolMetab 2016;101:841-6. [Google Scholar] |
| 2. | Goyal A, Panchani R, Varma T, Bhalla S, Tripathi S. Adrenal incidentaloma: A case of pheochromocytoma with sub-clinical Cushing’s syndrome. Indian J Endocrinol Metab 2013;17 (Suppl 1):S246-S248. [Google Scholar] |
| 3. | White A, Ray DW, Talbot A, Abraham P, Thody AJ, Bevan JS. Cushing’s syndrome due to phaeochromocytoma secreting the precursors of adrenocorticotropin. J Clin Endocrinol Metab 2000;85:4771-5. [Google Scholar] |
| 4. | Folkestad L, Andersen MS, Nielsen AL, Glintborg D. A rare cause of Cushing’s syndrome: An ACTH-secreting phaeochromocytoma. BM^ Case Rep 2014;2014:pii: bcr20142205487. [Google Scholar] |
| 5. | Chen H, Doppman JL, Chrousos GP, Norton JA, Nieman LK, Udelsman R. Adreno- corticotropic hormone-secreting pheochromocytomas: The exception to the rule. Surgery 1995;118:988-94. [Google Scholar] |
Fulltext Views
1,358
PDF downloads
11,399



