
Translate this page into:
Antiphospholipid anti-body syndrome presenting with diffuse alveolar haemorrhage and refractory autoimmune haemolytic anaemia
Correspondence to ROBIN CHOUDHARY; robinch19@gmail.com
[To cite: Marwah V, Hegde A, Adhikari S, Choudhary R. Antiphospholipid antibody syndrome presenting with diffuse alveolar haemorrhage and refractory autoimmune haemolytic anaemia. Natl Med J India 2024;37:329–31. DOI: 10.25259/NMJI_616_21]
Abstract
Antiphospholipid antibody syndrome (APS) is characterized by vascular thrombosis. Somewhat paradoxically, some patients with this disease develop diffuse alveolar haemorrhage. This bleeding is usually a delayed manifestation, occurring a few years after the onset of other manifestations and diagnosis of APS. We encountered a patient with an unusual presentation, i.e. onset with diffuse alveolar haemorrhage as well as vascular thromboses. He also had autoimmune haemolytic anaemia, which is infrequent in APS. The diagnosis of APS was based on elevated levels of IgM and IgG anti-cardiolipin antibody and IgG and IgM beta-2 GP1 antibodies. Treatment with high-dose glucocorticoids, anticoagulants and rituximab led to a decline in levels of these antibodies and was associated with a good and lasting clinical response.
INTRODUCTION
Antiphospholipid antibody syndrome (APS) is a systemic autoimmune disorder characterized by arterial and venous thrombosis and recurrent foetal loss. Hypercoagulable state and thromboembolic phenomenon are the most common mechanisms implicated in systemic, including pulmonary complications of primary APS. However, contrastingly, diffuse alveolar haemorrhage (DAH), which is a rare but potentially fatal complication of primary APS, occurs not due to this thromboembolic mechanism, but because of antiphospholipid antibody-mediated inflammation and destruction of arterioles, venules and alveolar septa. Autoimmune haemolytic anaemia (AIHA) is a common phenomenon in autoimmune diseases, although it has been rarely described in primary APS. Few case reports in the past have shown that there exists a lag period of 1 to 8 years from the diagnosis of APS to the first episode of DAH. We report a patient who presented with DAH and recurrent AIHA as the initial presentation of primary APS, which, to the best of our knowledge, is the first presentation of its kind in the contemporary literature.
THE CASE
A 30-year-old man of Indian origin, with no prior co-morbid conditions, presented to another hospital with complaints of intermittent high-grade fever associated with dry cough of three days duration. He developed dyspnoea on exertion (modified Medical Research Council Grade II), after a few days. On evaluation, he had leucocytosis with left shift and azotaemia. Chest X-ray showed bilateral hilar fluffy opacities (Fig. 1). High-resolution computed tomography (HRCT) of the chest revealed diffuse bilateral ground-glass opacities (GGOs; Fig. 2). The patient was diagnosed to have community-acquired pneumonia and treated with oxygen, antibiotics and supportive measures. Empiric antitubercular treatment was started on a clinicoradio-logical basis, and the patient was transferred to our centre.
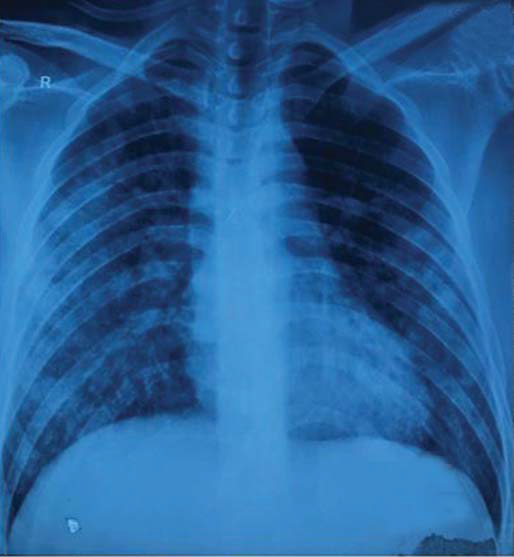
- Chest X-ray with ill-defined diffuse lung opacities suggestive of diffuse alveolar haemorrhage
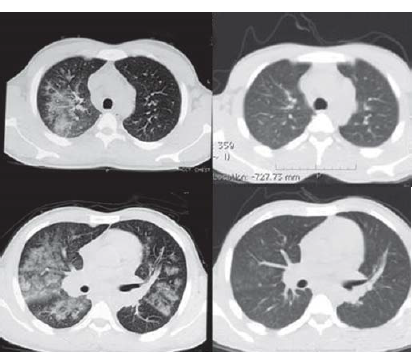
- (Left top and bottom) High resolution computed tomography of the chest showing patchy areas of ground glass opacities of bilateral upper, middle and lower lobes, sparing the periphery, suggestive of diffuse alveolar haemorrhage. (Right top and bottom) A repeat scan after 1 month of therapy shows notable resolution of diffuse alveolar haemorrhage
On presentation to our hospital, the patient appeared ill and had oliguria. He was febrile to touch, and on general examination had tachycardia, tachypnoea and hypoxaemia (SpO2 85% at room air, and 92% on 4 L of oxygen, given through nasal prongs). He had fine bilateral basal crepts on auscultation, while other systemic examinations including a dermatological and musculoskeletal system were normal. Laboratory parameters at admission revealed a drop in haemoglobin (from 15.0 g/dl to 6.2 g/dl), neutrophilic leucocytosis (total leucocyte count 19 000/cmm and polymorph 87%), thrombocytopenia (platelet <50 000/cmm) and features of acute kidney injury (urea/creatinine 182/5.7 mg/dl). Serum procalcitonin was normal. There were no active sediments, pus cells or microorganisms on urine examination. Blood and urine cultures were sterile. Sputum revealed haemosiderin-laden macrophages. With suspicion of diffuse alveolar haemorrhage (DAH), an autoimmune aetiology was looked for. Antinuclear antibody (ANA) and antineutrophil cytoplasmic antibody (ANCA), by immunofluorescence, were negative. Double-stranded DNA (ds DNA) and antiglomerular basement membrane antibody (anti-GBM Ab) were also negative. ELISA for human immunodeficiency virus (HIV), hepatitis B surface antigen (HBsAg) and hepatitis C serology were also negative. A peripheral blood smear (PBS) revealed an increase in microspherocytes, polychromatic red blood cells (RBCs) and presence of clumped RBCs with very low absolute platelet counts and large platelets. Serum lactate dehydrogenase (LDH) levels were also elevated (492 U/L, normal 280 U/L). The corrected reticulocyte count was 4.2%. Direct and indirect Coomb tests were strongly positive, and with evidence of extravascular haemolysis in PBS, along with an elevated reticulocyte count and LDH, led us to a diagnosis of autoimmune haemolytic anaemia (AIHA). The patient was managed with intravenous immunoglobulin (IVIg; total of 100 g divided over 5 days) as a life-saving measure. Continuous renal replacement therapy was also started for his acute kidney injury. The patient was transfused fresh blood, and an emergent bronchoscopy was done. The bronchoalveolar lavage (BAL) was neutrophil predominant (neutrophil 43% on differential count) with haemosiderin-laden macrophages (Fig. 3). No microorganisms could be identified on Gram and Ziehl Neelsen stain of the BAL fluid. The polymerase chain reaction test on the BAL fluid for mycobacteria was also negative, 2D echocardiography (2D echo) showed a large clot extending from the right common iliac vein up to the right atrium along with a thrombus in the right pulmonary artery (RPA) partially obstructing the lumen without any evidence of right heart failure.
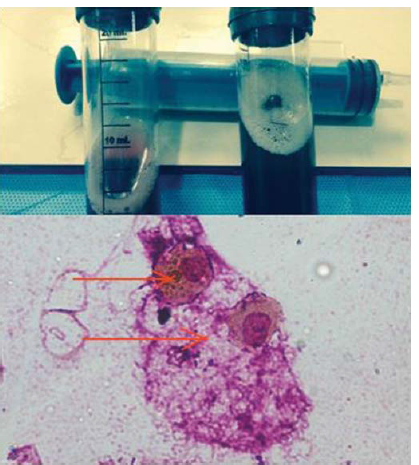
- Picture (top) showing bronchoalveolar lavage haemorrhagic, cytology micrograph (bottom; ×1000, Leishman Giemsa stain) reveals haemosiderin-laden macrophages (red arrow)
As the patient presented with both, haemorrhagic and prothrombotic manifestations in the background of recurrent AIHA, and thrombocytopenia with autoimmune markers, the patient was investigated further. His anti-cardiolipin antibody (aCL) IgG and IgM by the enzyme-linked immunosorbent assay (EIA) was 28.20 GPL (cut-off <15 GPL) and 90.67 MPL (cut-off 12.50 MPL), respectively. Beta 2 GP1 antibody IgG and IgM by EIA were also significantly raised, the titre being 28.69 SGU (cutoff <20 SGU) and 65.83 SMU (cut-off <20 SMU), respectively. The lupus anticoagulant by diluted Russell viper venom time was negative. The patient was finally diagnosed as primary APS, with DAH and AIHA.
He was managed with methylprednisolone (1 g daily for 3 days), followed by maintenance therapy with oral prednisolone, 40 mg daily. Anticoagulation was initiated with low molecular weight heparin (LMWH). His repeat 2D echo showed regression of the clot in the RPA and Fondaparinux was switched to warfarin, with a target INR of 2–3. His steroid tapering was commenced. However, his haemoglobin again decreased to 5 g/dl with recurrence of features of haemolysis on PBS. Given the refractory nature of AIHA in the background of APS, B-cell depletion with Rituximab was considered. The patient was given rituximab 500 mg weekly, for a total of four doses. A repeat testing of aCL and Beta2 GP1antibodies at the end of 12 weeks, revealed falling titres. However, there were no fresh episodes of DAH, thrombotic events or AIHA on follow-up.
DISCUSSION
APS is a systemic autoimmune disorder characterized by arterial and venous thrombosis and recurrent foetal loss. It can be associated with thrombocytopenia accompanied by elevation of at least one of the antiphospholipid (aPL) antibodies, measured on two or more occasions at least 12 weeks apart.1 It was first recognized in patients with systemic lupus erythematosus (SLE) and later found in association with other autoimmune disorders, malignancies and infections including HIV. This condition has also been recognized as a syndrome, that can develop independently of any underlying disease, known as primary APS. Frequent pulmonary complications associated are acute pulmonary thromboembolism, chronic thromboembolic pulmonary hypertension, pulmonary microthrombosis and fibrosing alveolitis.2 Although the thromboembolic phenomenon is the most common mechanism causing pulmonary complications in primary APS, rarely it can present as DAH.2,3
DAH is a rare and life-threatening complication of primary APS. Bleeding into the alveolar spaces of the lungs characterizes the syndrome of DAH and is due to disruption of the alveolar– capillary basement membrane which is caused by injury or inflammation of the arterioles, venules or alveolar septal capillaries. Although the aetiology of primary APS-associated DAH has been linked to autoantibodies, the precise pathogenic mechanisms have not been fully elucidated. It has been suggested that primary APS-associated DAH does not represent a thrombotic complication, but an inflammatory process precipitated by the circulating aPL.3,4 Deane and West hypothesized that capillaritis in primary APS-associated DAH is due to aPL-induced upregulation of endothelial cell adhesion molecules (ECAMs) with subsequent neutrophil recruitment and migration into the alveolar septa resulting in tissue destruction and haemorrhage.5
There are few case reports of DAH in primary APS. A retrospective study of 18 cases from Mayo Clinic reported the median time of diagnosis of APS to the first episodes of DAH was 5.9 years, earliest being 1.4 years.2 However, in our patient DAH was the initial manifestation, which was confirmed by the presence of GGO in HRCT, drop in haemoglobin and presence of haemosiderin-laden macrophages in BAL. Further, neutrophilia of BAL was consistent with the hypothesis of Deane and West.5 The absence of markers of connective tissue disorder and vasculitis such as ANA, ds DNA, ANCA and Anti GBM Ab in our patient ruled out the simultaneous presence of any systemic autoimmune disease.
The mainstay of management is oral or parenteral corticosteroids, depending on the severity of the disease.2 There is no firm recommendation regarding steroid-sparing immunosuppressants.6 In a retrospective study of 18 patients from Mayo Clinic, cyclophosphamide and rituximab were the most used immunosuppressants and showed favourable results.2 Our patient received 1 g of parenteral methylpredniso-lone for three days, followed by oral prednisolone 40 mg and showed dramatic improvement in the form of resolution of dyspnoea.
Our patient had a large clot in the inferior vena cava, right atrium and the RPA, which could have led to life-threatening pulmonary thromboembolism. Therefore, LMWH was carefully initiated for the prevention of further complications. A review of the literature suggests that heparin has a positive role not only in treating thrombosis but also in DAH. Girardi et al. proposed that heparin blocks the activation of complement initiated by aPL antibody by inhibiting C1q and interference with C4, thus preventing the formation of C3 amplification convertase.7,8
Our patient had a high titre of circulating auto-antibodies leading to immune-complex mediated haemolytic anaemia and thrombocytopenia. Hence, we decided to start him on IVIg as its immuno-modulatory and anti-inflammatory effects target many pathways in the innate and adaptive immunity systems.9
Rituximab is a chimeric monoclonal antibody targeting the CD 20 antigen on B-cell surface.10,11 A randomized trial by Birgens et al. compared glucocorticoid monotherapy with glucocorticoid and rituximab combination in patients with AIHA and concluded that relapse-free survival was better after combination therapy, than with glucocorticoid monotherapy alone. They suggested that rituximab-glucocorticoid combination could be the first-line management in such cases.12
The repeat testing for aPL at 12 weeks showed decreasing level of titres of Beta 2 GP1 IgG and IgM, which supported the hypothesis that Beta 2 GP1 is the pathogenic antibody in primary APS-associated DAH.11
Conclusion
DAH and AIHA are rare and potentially life-threatening complications of primary APS, and seldom can be the presenting manifestations of APS. The treating physician needs to have a high index of suspicion for these entities, given the high morbidity. The diagnosis of DAH should be suspected in a patient of known APS when presenting with dyspnoea, falling haemoglobin and alveolar filling pattern on the chest X-ray. Fibreoptic bronchoscopy and BAL are valuable in establishing the diagnosis of DAH and in ruling out an infectious aetiology. Prompt recognition, with optimum immunosuppression and adequate anticoagulation are the key factors in preventing morbidity and mortality.
Conflicts of interest
None declared
References
- International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS) J Thromb Haemost. 2006;4:295-306.
- [CrossRef] [PubMed] [Google Scholar]
- Primary antiphospholipid syndrome-associated diffuse alveolar hemorrhage. Arthritis Care Res (Hoboken). 2014;66:301-10.
- [CrossRef] [PubMed] [Google Scholar]
- Primary antiphospholipid syndrome associated with diffuse alveolar hemorrhage and pulmonary thromboembolism. Intern Med. 2015;54:2029-33.
- [CrossRef] [PubMed] [Google Scholar]
- Pulmonary manifestations of the antiphospholipid antibody syndrome. Clin Chest Med. 2010;31:537-45.
- [CrossRef] [PubMed] [Google Scholar]
- Antiphospholipid antibodies as a cause of pulmonary capillaritis and diffuse alveolar hemorrhage: A case series and literature review. Semin Arthritis Rheum. 2005;35:154-65.
- [CrossRef] [PubMed] [Google Scholar]
- Antiphospholipid antibodies-associated diffuse alveolar hemorrhage. Semin Arthritis Rheum. 2015;44:652-7.
- [CrossRef] [PubMed] [Google Scholar]
- Complement C3 activation is required for antiphospholipid antibody-induced fetal loss. J Exp Med. 2002;195:211-20.
- [CrossRef] [PubMed] [Google Scholar]
- Heparin prevents antiphospholipid antibody-induced fetal loss by inhibiting complement activation. Nat Med. 2004;10:1222-6.
- [CrossRef] [PubMed] [Google Scholar]
- Mechanisms of action and historical facts on the use of intravenous immunoglobulins in systemic lupus erythematosus. Autoimmun Rev. 2019;18:279-86.
- [CrossRef] [PubMed] [Google Scholar]
- Efficacy and safety of rituximab in autoimmune and microangiopathic hemolytic anemia: A systematic review and meta-analysis. Exp Hematol Oncol. 2020;9:6.
- [CrossRef] [PubMed] [Google Scholar]
- Rituximab induces resolution of recurrent diffuse alveolar hemorrhage in a patient with primary antiphospholipid antibody syndrome. Lupus. 2012;21:438-40.
- [CrossRef] [PubMed] [Google Scholar]
- A phase III randomized trial comparing glucocorticoid monotherapy versus glucocorticoid and rituximab in patients with autoimmune haemolytic anaemia. Br J Haematol. 2013;163:393-9.
- [CrossRef] [PubMed] [Google Scholar]



