Translate this page into:
Association of presenile cataract with galactose-1-phosphate uridyl transferase gene mutations
2 Central Research Laboratory, Sri Aurobindo Medical College and PG Institute, Indore Ujjain Highway, Indore, Madhya Pradesh, India
Corresponding Author:
Ravindra Kumar
Central Research Laboratory, Sri Aurobindo Medical College and PG Institute, Indore Ujjain Highway, Indore, Madhya Pradesh
India
ravindrachhabra@gmail.com
| How to cite this article: Nema N, Kumar R, Verma A, Verma S, Chaturvedi K. Association of presenile cataract with galactose-1-phosphate uridyl transferase gene mutations. Natl Med J India 2017;30:73-75 |
Abstract
Background. Presenile cataract is commonly idiopathic in origin. However, patients with presenile cataract could have an underlying genetic abnormality of galactose metabolism. We studied the association, if any, between idiopathic presenile cataract and galactose-1 -phosphate uridyl transferase (GALT) gene mutation.Methods. We selected 50 patients with idiopathic presenile cataract, <45 years of age, and 50 age- and sex-matched controls for the study. Mutations in the GALT gene were determined by polymerase chain reaction restriction fragment length polymorphism. The classical galactosaemia was characterized by Q188R and K285N mutations, whereas Duarte galactosaemia by N314D mutations (Duarte-2: N314D with IVS5-24G >A and Duarte-1: N314D without IVS5- 24G>A).
Results. The most common mutation observed was the N314D (Duarte) mutation. The frequencies of classical and N31 4D alleles in patients with presenile cataract (16%) and controls (26%) were not statistically different (p=0.32, OR 0.54, 95% CI 0.20–1.45). Similarly, there was no statistically significant difference in the frequency distribution of Duarte-1 (p=0.77, OR 0.77, 95% CI 0.23–0.24) and Duarte-2 (p=0.44, OR 0.38, 95% CI 0.07–2.03) galactosaemia mutations in patients and controls.
Conclusion. Duarte galactosaemia, a milder form of the disease, is more common than classical galactosaemia in the Indian population. Duarte galactosaemia is unlikely to be a causative factor in presenile cataract.
Introduction
Cataract is an opacity in the natural crystalline lens that results from change in the refractive index of the lens.[1] A congenital cataract occurs within the first year of life, a juvenile cataract within the first decade of life, a presenile cataract before the age of 45 years, and senile cataract, thereafter.[2] The age of onset of cataract does not necessarily indicate its aetiology.[3]
There is an association between partial galactose metabolism impairment and idiopathic presenile cataracts.[4],[5],[6] Galactose accumulation in tissues is due to deficient galactose-1 -phosphate uridyl transferase (GALT) activity. The classical galactosaemia is caused by mutation (Q188R and K285N) in the GALT gene located on chromosome 9p13.[7] The homozygotes have no significant GALT activity and the condition is usually lethal.[8] Heterozygotes have 50% residual GALT activity and are clinically asymptomatic. Another form of GALT deficiency occurs due to mutation of N314D, which is characteristic of Duarte-1 and -2 variants of galactosaemia. Duarte-2 variant has N314D mutation in three intronic variations: IVS4-27G>C (G1105C); IVS5+62G>A (G1323A) and IVS5-24G>A (G1391A) along with a deletion in the 5' promoter region (–119/–116delGTCA) of the GALT gene. In Duarte-1 (or Los Angles, LA) variant a silent mutation of L218L is present together with N314D mutation.[9],[10] Duarte-1 and -2 variants have variable GALT activity and are at increased risk for presenile cataract formation.[11],[12],[13],[14]
Cataract, a common cause of blindness in India, occurs at an earlier age than in western countries.[15],[16],[17],[18] Most studies have found no cause for the development of presenile cataract.[19],[20],[21] Many of these idiopathic presenile cataracts could be attributed to the GALT gene mutation that results in decreased GALT activity, high galactose concentration and cataract formation.[4],[5],[6],[13] Therefore, we ascertained the prevalence of the GALT gene mutation in patients with idiopathic presenile cataract.
Methods
We selected 50 patients with idiopathic presenile cataract (age <45 years) visiting the Department of Ophthalmology, at our tertiary care centre. The study was approved by the institutional ethical committee and followed the tenets of the Declaration of Helsinki.
Patients with a history of atopy or allergy, ocular trauma, prolonged topical or systemic medication, intraocular surgery, recurrent red eye, diabetes mellitus and smoking were excluded from the study. An equal number of age- and sex-matched healthy controls with normal vision and with no previous history of cataract surgery were recruited as controls.
Clinical examination
The presence of cataract was confirmed on slit-lamp examination of the anterior segment of eye after pupillary dilatation. Any opacity in the crystalline lens was designated as cataract and it was classified as cortical, nuclear and/or posterior subcapsular. The axial length of the eyeball was measured using an ultrasonic biometer. Patients with an axial length >26 mm were labelled as high myopic and excluded from the study.
Laboratory investigations
After obtaining written informed consent, 2 ml of peripheral venous blood was collected in an EDTA tube. DNA was isolated using QIAamp DNA Mini Kit (Qiagen) following the instruction manual. Classical galactosaemia mutations (Q188R and K285N) and Duarte galactosaemia mutations (Duarte-2: N314D with IVS5-24G>A; Duarte-1: N314D lacking IVS5-24G>A) were identified by polymerase chain reaction restriction fragment length polymorphism (PCR-RFLP). The obtained PCR product was characterized for Q188R, K285N, N314D mutations and IVS1- 24G>A variant based on the observation that all these putative mutations created an additional restriction site for the HpaII, Tsp509I, and AvaII restriction endonuc lease, respectively, whereas intronic variation IVS5-24G>A (1391G>A) eliminated restriction site for the endonuclease SacI that was present in the normal gene.
PCR products were digested with 3U of corresponding restriction enzymes overnight at 37 °C to determine the mutation. Products were separated on 2% agarose gel electrophoresis. The expected band pattern showed the presence or absence of mutation.
Duarte allelic genotype was attributed to alleles containing the N314D mutation and the IVS5-24G>A intronic variation. Alleles with the N314D mutation along with the IVS5-24G>A variation were labelled as Duarte-2, and if IVS5-24G>A variation was not there, the GALT genotype was labelled as Duarte-1 variant.
Statistical analysis
The chi-square test was used to assess the difference in frequency of genotypes in patients and controls. The Mann–Whitney U test was used for differences of quantitative data between the two groups.
Results
The median age of patients and controls was 36 (31–45) years and 34 (31–42) years, respectively. The male-to-female ratio in patients and controls was 23:27 and 31:19, respectively. There was no significant difference in age (p =0.1) and sex (p=0.11) of patients and controls.
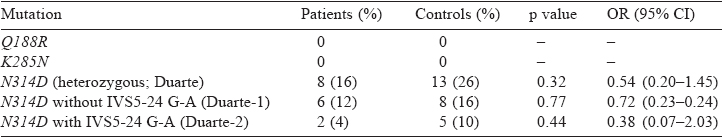
No mutant allele for Q188R and K285 Nmutations was observed in either the patients or controls. The N314D mutation was present in 8 patients and 13 controls. The frequency of N314 D in controls followed the Hardy-Weinberg equilibrium.
The frequency distribution of N314D in patients was not significantly different in controls (16% v. 26%; p=0.32, OR 0.54, 95% CI 0.20–1.45). Of 8 patients and 13 controls who were positive for N314D mutation, 2 patients and 5 controls also had the IVS5-24G>A polymorphism and thus belonged to the Duarte- 2 galactosaemia category [Table - 1]. The remaining N314D-positive patients were considered as Duarte-1 galactosaemia. No statistical significant difference was observed between the patients and controls in the Duarte-2 allele frequencies (4% v. 10%; p=0.24) and the Duarte-1 or Los Angeles allele frequencies (12% v. 16%, p=0.77). No individual was found to be homozygous for the Duarte-1 or -2 mutation.
![[Table - 1]](#tbl_NatlMedJIndia_2017_30_2_73_212908_t1.jpg){kind=link}
Discussion
About 80% of blindness in India is because of cataract.[15],[16] An earlier age of onset of cataract in the Indian population suggests either an enormous exposure to environmental risk factors or a genetic predisposition to cataract formation.[22],[23],[24] Presenile cataract refers to onset that occurs at any time from early adult life to 45 years of age.[21] The common causes for the development of presenile cataract include atopy (dermatitis, asthma), high myopia, diabetes mellitus, long-term use of corticosteroids in any form, family history of early cataract and idiopathic.[19],[21],[25],[26] Pregnancy, dehydration, trauma, exposure to radiations and tobacco use are also reported to be associated with presenile cataract.[23],[24],[27],[28]
Galactosaemia, similar to diabetes mellitus, is a metabolic disorder that causes improper galactose metabolism and a predisposition to cataract formation.[5],[29] This genetic inborn error of metabolism is caused by mutation in genes that involve enzymes needed for galactose breakdown in the body. The most common and severe form is classical galactosaemia due to mutation in the GALT gene. The other less common types are galactosaemia type II due to mutation in the GALK1 (galactokinase) gene and galactosaemia type III due to mutation in the GALE (galactose epimerase) gene.
In galactosaemia, early and presenile cataract formation occurs due to deficient GALT enzyme. This leads to accumulation of galactose in the lens, which is reduced to galactitol by aldose reductase enzyme. Galactitol causes osmotic swelling of the lens resulting in cataract formation. However, the severity of clinical features in a galactosaemic patient depend upon the residual GALT enzyme activity. In classical galactosaemia homozygotes having no GALT activity, the condition is most severe and lethal.[8] On the contrary, heterozygotes and Duarte galactosaemia show 50% or more GALT activity and are, therefore, asymptomatic at birth. Nonetheless, studies have shown that these galactosaemic mutants who appear normal at birth have an increased risk for developing idiopathic presenile cataract later in life.[4],[5],[6],[13],[29]
In western countries, the diagnosis of galactosaemia is usually made by newborn screening tests. Such screening is not done in India.[30] The early diagnosis of disease helps in its efficient management. Therefore, this study was done to find a relation, if any, between the GALT gene mutation and idiopathic presenile cataract formation. To the best of our knowledge, this is the first study of its kind in the Indian population.
The DNA analysis was done to demonstrate mutations in the GALT gene in patients with presenile cataract. The two most common mutations, Q188R and K285N, with almost no GALT enzyme activity result in classical galactosaemia in Caucasians.[31] A higher K285N mutation was seen in patients of presenile cataract than in the control group.[5],[12],[32] Interestingly, these mutations were found to be completely missing from both the patients and controls in our study [Table - 1]. A lower frequency of Q188R mutation has been reported in Indian galactosaemic patients as compared to American and European populations.[33]
The Los Angeles allele (N314D) or Duarte-1 galactosaemia was present with a frequency of 12% and 16% in patients and controls, respectively in our population [Table - 1]. A higher frequency of Duarte variant was reported by Singh et al. and they suggested that Indians suffer from a milder form of galactosaemia.[33] Among the non-Caucasian populations, the highest frequency of N314D mutation was in Indians.[12] Similarly, an increased frequency of this variant has also been reported from other parts of the world—the Slovenian general population showed a frequency of 5%–15%, Caucasians 8.8% and Germans 14.9%.[31] However, we found no statistically significant difference in the occurrence of the mutation between patients with idiopathic presenile cataract and controls.
There are certain limitations of our study. We have not assessed the GALT activity in patients and controls. Second, we have not sequenced the GALT gene to rule out the possibility of other mutations. Third, the sample size of the study is small to arrive at a definitive conclusion. Therefore, studies with a larger sample size will be required to corroborate our findings.
In conclusion, we believe that defects in the GALT gene may not have a role in the formation of presenile cataract.
| 1. | Benedek GB. Theory of transparency of the eye. Appi Opt 1971; 10: 459–73. [Google Scholar] |
| 2. | Shiels A, Hejtmancik JF. Genetics of human cataract. Clin Genet 2013; 84: 120–7. [Google Scholar] |
| 3. | Hejtmancik J, Datilles M. Congenital and inherited cataracts. In: Tasman W (ed). Duane's clinical ophthalmology. Volume 1. Philadelphia, PA:J.B. Lippincott Co. and University of Chicago; 1994. [Google Scholar] |
| 4. | Stevens RE, Datiles MB, Srivastava SK, Ansari NH, Maumenee AE, Stark WJ. Idiopathic presenile cataract formation and galactosaemia. Br J Ophthalmol 1989; 73: 48–51. [Google Scholar] |
| 5. | Karas N, Gobec L, Pfeifer V, Mlinar B, Battelino T, Lukac-Bajalo J. Mutations in galactose-1-phosphate uridyl transferase gene in patients with idiopathic presenile cataract. J Inherit Metab Dis 2003; 26: 699–704. [Google Scholar] |
| 6. | Karas-Kuzelicki N, Pfeifer V, Lukac-Bajalo J. Synergistic effect of high lactase activity genotype and galactose-1-phosphate uridyl transferase (GALT) mutations on idiopathic presenile cataract formation. Clin Biochem 2008; 41: 869–74. [Google Scholar] |
| 7. | Shih LY, Suslak L, Rosin I, Searle BM, Desposito F. Gene dosage studies supporting localization of the structural gene for galactose-1-phosphate uridyl transferase (GALT) to band p13 of chromosome 9. Am J Med Genet 1984; 19: 539–43. [Google Scholar] |
| 8. | Segal S, Berry GT. Disorders of galactose metabolism. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds). The metabolic and molecular bases of inherited disease, Vol. 1. 7th ed. New York:McGraw-Hill; 1995:967–1000. [Google Scholar] |
| 9. | Podskarbi T, Kohlmetz T, Gathof BS, Kleinlein B, Bieger WP, Gresser U, et al. Molecular characterization of Duarte-1 and Duarte-2 variants of galactose-1-phosphate uridyl transferase. J Inherit Metab Dis 1996; 19: 638–44. [Google Scholar] |
| 10. | Trbusek M, Francová H, Kozák L. Galactosemia: Deletion in the 5' upstream region of the GALT gene reduces promoter efficiency. Hum Genet 2001; 109: 117–20. [Google Scholar] |
| 11. | Tyfield L, Reichardt J, Fridovich-Keil J, Croke DT, Elsas LJ 2nd, Strobl W, et al. Classical galactosemia and mutations at the galactose-1 -phosphate uridyl transferase (GALT) gene. Hum Mutat 1999; 13: 417–30. [Google Scholar] |
| 12. | Suzuki M, West C, Beutler E. Large-scale molecular screening for galactosemia alleles in a pan-ethnic population. Hum Genet 2001; 109: 210–15. [Google Scholar] |
| 13. | Skalka HW, Prchal JT. Presenile cataract formation and decreased activity of galactosemic enzymes. Arch Ophthalmol 1980; 98: 269–73. [Google Scholar] |
| 14. | Vaca-Pacheco G, Medina C, García-Cruz D, Sánchez-Corona J, Chávez-Anaya E, Jaimes C, et al. Identification of inborn errors of galactose metabolism in patients with cataracts. Arch Invest Med (Mex) 1990; 21: 127–32. [Google Scholar] |
| 15. | Mohan M. Survey of blindness—India (1986–1989). In: Summary results: Programme for the control of blindness. New Delhi:Ministry of Health and Family Welfare, Government of India; 1992. [Google Scholar] |
| 16. | Dandona L, Dandona R, Srinivas M, Giridhar P, Vilas K, Prasad MN, et al. Blindness in the Indian state of Andhra Pradesh. Invest Ophthalmol Vis Sci 2001; 42: 908–16. [Google Scholar] |
| 17. | Chatterjee A, Milton RC, Thyle S. Prevalence and aetiology of cataract in Punjab. Br J Ophthalmol 1982; 66: 35–42. [Google Scholar] |
| 18. | Thompson JR. The demand incidence of cataract in Asian immigrants to Britain and their descendants. Br J Ophthalmol 1989; 73: 950–4. [Google Scholar] |
| 19. | Praveen MR, Shah GD, Vasavada AR, Mehta PG, Gilbert C, Bhagat G. A study to explore the risk factors for the early onset of cataract in India. Eye (Lond) 2010; 24: 686–94. [Google Scholar] |
| 20. | Rahman A, Yahya K, Shaikh A, Fasih U, Zuberi BF. Risk factors associated with presenile cataract. Pak J Med Sci 2011; 27: 145–8. [Google Scholar] |
| 21. | Tsai CK, Teng MC, Wu PC, Kuo HK. Clinical features of patients featuring cataracts in a myopia-endemic area of Taiwan. Chang GungMed J 2006; 29: 406–11. [Google Scholar] |
| 22. | Mohan M, Sperduto RD, Angra SK, Milton RC, Mathur RL, Underwood BA, et al India–US case–control study of age-related cataracts. India–US Case–Control Study Group. Arch Ophthalmol 1989; 107: 670–6. [Google Scholar] |
| 23. | Minassian DC, Mehra V, Verrey JD. Dehydrational crises: A major risk factor in blinding cataract. Br J Ophthalmol 1989; 73: 100–5. [Google Scholar] |
| 24. | Minassian DC, Mehra V, Jones BR. Dehydrational crises from severe diarrhoea or heatstroke and risk of cataract. Lancet 1984; 7: 751–3. [Google Scholar] |
| 25. | Gopalakrishna K, Rao PN, Nayak BR, Pattabiraman TN. Nonenzymatic glucosylation of lens proteins in different types of cataracts. Indian J Med Res 1983; 78: 426–30. [Google Scholar] |
| 26. | Mitry D, Singh J, Kiire CA, Hegde V. Atopic dermatitis, facial trauma, and cataract surgery. Can J Ophthalmol 2009; 44: 716. [Google Scholar] |
| 27. | Minassian DC, Mehra V, Reidy A. Childbearing and risk of cataract in young women: An epidemiological study in central India. Br J Ophthalmol 2002; 86: 548–50. [Google Scholar] |
| 28. | Bhatnagar R, West Jr KP, Vitale S, Sommer A, Joshi S, Venkataswamy G. Risk of cataract and history of severe diarrheal disease in southern India. Arch Ophthalmol 1991; 109: 696–9. [Google Scholar] |
| 29. | Lukac Bajalo J. Idiopathic presenile cataracts and galactosemia. Adv Clin Pathol 1997; 1 (Suppl 1):L5. [Google Scholar] |
| 30. | Mathew V, Ramakrishnan A, Srinivasan R, Sushma K, Bantwal G, Ayyar V. Erroneous glucose recordings while using mutant variant of quinoprotein glucose dehydrogenase glucometer in a child with galactosemia. Indian J Endocrinol Metab 2013; 17 (Suppl 1):S289–S291. [Google Scholar] |
| 31. | Lukac-Bajalo J, Marc J, Mlinar B, Karas N, Krzisnik C, Battelino T. Frequencies of Q188R and N314D mutations and IVS5-24g>A intron variation in the galactose-1- phosphate uridyl transferase (GALT) gene in the Slovenian population. Clin Chem Lab Med 2002; 40: 1109–13. [Google Scholar] |
| 32. | Lukac-Bajalo J, Kuzelicki NK, Zitnik IP, Mencej S, Battelino T. Higher frequency of the galactose-1-phosphate uridyl transferase gene K285N mutation in the Slovenian population. Clin Biochem 2007; 40: 414–15. [Google Scholar] |
| 33. | Singh R, Thapa BR, Kaur G, Prasad R. Frequency distribution of Q188R, N314D, Duarte 1, and Duarte 2 GALT variant alleles in an Indian galactosemia population. Biochem Genet 2012; 50: 871–80. [Google Scholar] |
Fulltext Views
1,074
PDF downloads
279



