Translate this page into:
Haemoglobin H disease: All in the family
Corresponding Author:
Jayashree D Kulkarni
Department of Pathology and Laboratory Medicine, Columbia Asia Referral Hospital Yeshwanthpur, Bengaluru, Karnataka
India
jayashreedk@gmail.com
| How to cite this article: Kulkarni JD. Haemoglobin H disease: All in the family. Natl Med J India 2016;29:310 |
An 18-year-old man had persistent anaemia not responding to iron and vitamin B12 supplements, and splenomegaly. Investigations showed a haemoglobin level of 6.7 g/dl, mean corpuscular volume (MCV) of 70.2 fL, mean corpuscular haemoglobin of 18.0 pg/dl, iron studies were normal and the lactate dehydrogenase level was 243 i.u./L. The peripheral smear showed severe microcytic hypochromic anaemia with polychromasia. The histogram of red blood cell (RBC) size showed a marked shift to the left and a high take-off from the Y-axis ([Figure - 1]). We suspected thalassaemia or haemoglobinopathy. High-performance liquid chromatography (HPLC) done on Biorad D-10 revealed two fast peaks at <0.65 minute retention time, indicating haemoglobin H (HbH) disease ([Figure - 2]). Methylene blue stained peripheral smear showed a golf ball appearance of the erythrocytes with numerous HbH inclusions, indicative of HbH disease ([Figure - 3]). We recommended screening of the family members. The mother had RBC indices suggestive of a thalassaemia carrier, normal HPLC but occasional HbH inclusions. The father′s values were normal. The brother′s RBC histogram, HPLC and methylene blue-stained smear were similar to that of our patient.
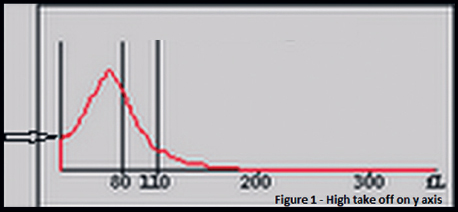 |
| Figure 1. Red blood cell histogram showing high take off on Y-axis |
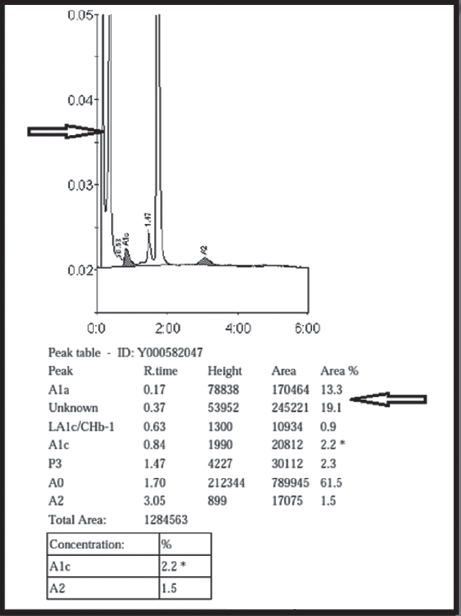 |
| Figure 2. High-performance liquid chromatogram showing two fast peaks |
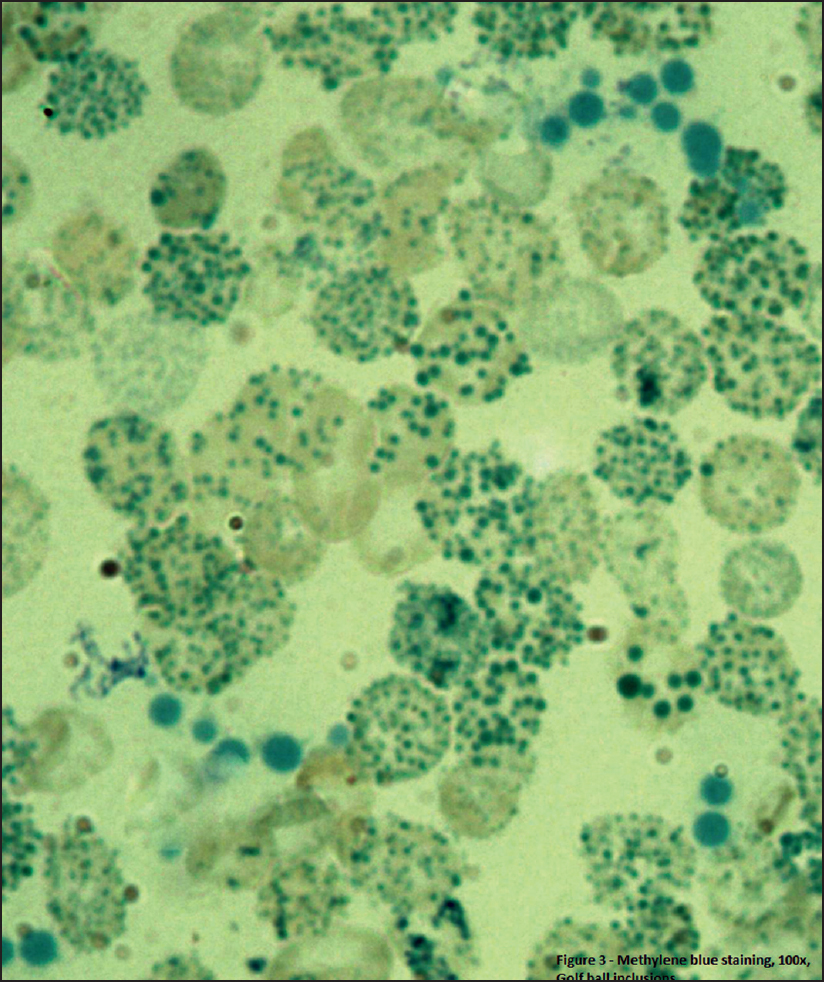 |
| Figure 3. Erythrocytes with a golf ball appearance and inclusions (Methylene blue, 100ƒs) |
Detection of alpha thalassaemia is important because it is common, but may remain silent or can manifest with haemolysis in HbH disease (alpha thalassaemia with three chain deletion), and have implications for genetic counselling. The RBC histogram is of crucial importance in diagnosis. Methylene blue stain may help in cases with low MCV, suggestive of thalassaemia silent carrier, if the haemoglobin A2 is normal or reduced, especially when patients cannot afford expensive confirmatory tests.
Fulltext Views
1,580
PDF downloads
532




![[Figure - 1]](#fig_NatlMedJIndia_2016_29_5_310_197828_f1.jpg){kind=link}
![[Figure - 2]](#fig_NatlMedJIndia_2016_29_5_310_197828_f2.jpg){kind=link}
![[Figure - 3]](#fig_NatlMedJIndia_2016_29_5_310_197828_f3.jpg){kind=link}