Translate this page into:
Hereditary haemorrhagic telangiectasia
Corresponding Author:
Mayank Jain
Institute of GI Sciences and Research, Gleneagles Global Health City, Chennai, Tamil Nadu
India
mayank4670@rediffmail.com
| How to cite this article: Tapadia A, Mahadevan B, Jain M, Sameer Kumar G S, Venkataraman J. Hereditary haemorrhagic telangiectasia. Natl Med J India 2020;33:60 |
Hereditary haemorrhagic telangiectasia (HHT), also known as Rendu–Osler–Weber disease, is an autosomal dominant disorder of the fibrovascular tissue.[1] It is characterized by the classical triad of mucocutaneous telangiectasia, arteriovenous malformations with recurrent epistaxis and haemorrhages.[2]
A 60-year-old female presented with a history of passage of intermittent black tarry stools in the preceding 10 years requiring multiple blood transfusions and intravenous iron preparations. There was a history of recurrent episodes of epistaxis since childhood requiring nasal packs. Her elder sibling and daughter had a similar history of recurrent epistaxis and transfusion-dependent anaemia. On examination, she was pale with punctuate tiny macules on the fingertips of both hands, tongue and lips [Figure - 1]a and [Figure - 1]b. Nasal endoscopy showed multiple nasal angiodysplasias [Figure - 1]c. Oesophagogastroduodenoscopy revealed multiple angiodysplasias in the stomach and the duodenum [Figure - 1]d. Colonoscopy was normal. Capsule enteroscopy showed oozing of blood from angiodysplastic lesions in the jejunum. Rest of the small bowel was normal. Argon plasma coagulation was done for the bleeder using balloon enteroscopy. Computed tomography of the abdomen with an angiogram showed aneurysmal dilatation of the portal vein with multiple venovenous malformations [Figure - 1]e. Two-dimensional echocardiogram showed moderate pulmonary arterial hypertension. Based on Curacao criteria,[3] the patient fulfilled the definitive criteria for the diagnosis of HHT, i.e. autosomal mode of inheritance, telangiectasia of the fingertips, spontaneous and recurrent epistaxis and gastrointestinal bleed. She was managed initially with lanreotide and later with thalidomide without much benefit. The patient is currently on symptom-based supportive care.
![[Figure - 1]](#fig_NatlMedJIndia_2020_33_1_60_308260_f1.jpg){kind=link}
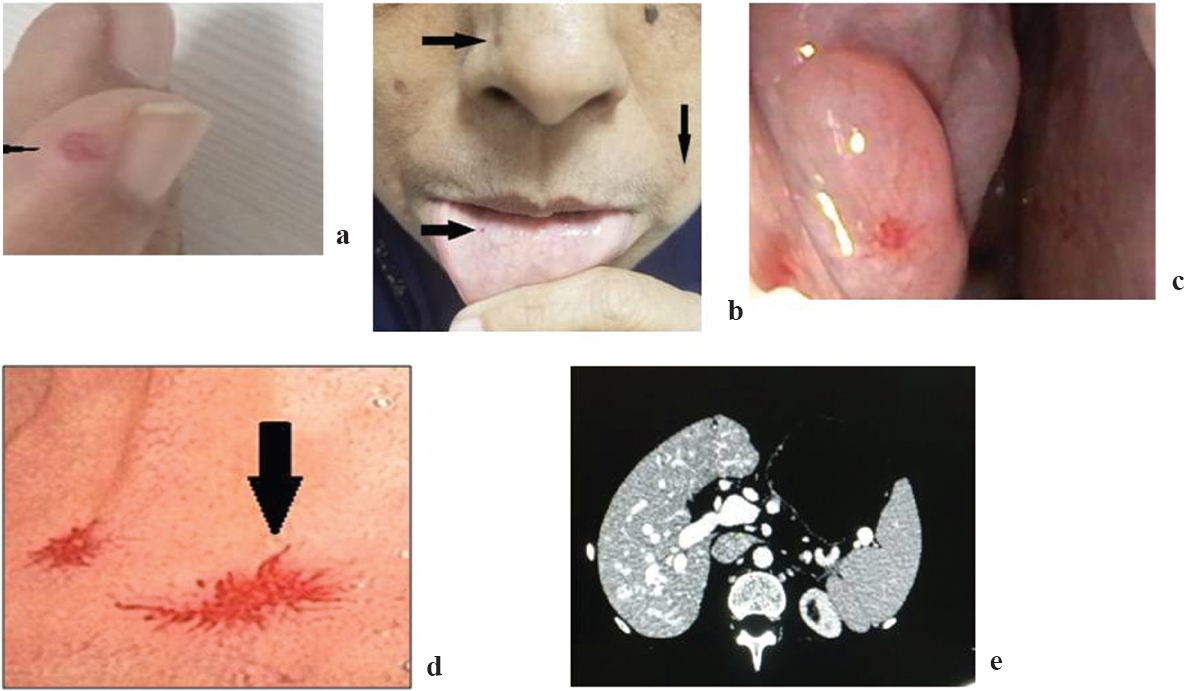 |
| Figure 1: (a) Telangiectasia on the fingertip; (b) telangiectasia on buccal mucosa, cheek and nose; (c) telangiectasia in the nasal cavity; (d) duodenal telangiectasias; (e) computed tomography of liver showing venovenous malformations |
Conflicts of interest. Nil
| 1. | te Veldhuis EC, te Veldhuis AH, van Dijk FS, Kwee ML, van Hagen JM, Baart JA, et al. Rendu–Osler–Weber disease: Update of medical and dental considerations. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2008;105:e38–e41. [Google Scholar] |
| 2. | McDonald J, Bayrak-Toydemir P, Pyeritz RE. Hereditary hemorrhagic telangiectasia: An overview of diagnosis, management, and pathogenesis. Genet Med 2011;13:607–16. [Google Scholar] |
| 3. | Shovlin CL, Guttmacher AE, Buscarini E, Faughnan ME, Hyland RH, Westermann CJ, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu–Osler–Weber syndrome). Am J Med Genet 2000;91:66–7. [Google Scholar] |
Fulltext Views
2,141
PDF downloads
1,917



