
Translate this page into:
Lamellar cataract in a child with Alagille syndrome
Correspondence to DEEKSHA RANI; deekshamedico@gmail.com
[To cite: Khokhar S, Rani D, Namdev V, Rathod A, Kumar S. Lamellar cataract in a child with Alagille syndrome. Natl Med J India 2024;37: 327–8. DOI: 10.25259/NMJI_761_2022]
Abstract
Alagille syndrome is a multisystem disorder inherited in an autosomal dominant manner with a variable phenotypic presentation. Typical features include intrahepatic bile duct paucity, butterfly-shaped vertebrae, typical facies, axenfeld anomaly (posterior embryotoxon) and cardiac abnormalities. Since this syndrome has typical ocular associations, ophthalmologists also have an important role in diagnosing the condition. Ocular features include posterior embryotoxon, corneal pannus, chorioretinal abnormalities and posterior subcapsular cataract. We report a toddler, diagnosed with Alagille syndrome who presented to us with a visually significant lamellar cataract in both eyes. To the best of our knowledge, this is the first case reporting lamellar cataract in a toddler with Alagille syndrome.
INTRODUCTION
First described by a French paediatrician, Daniel Alagille in 1969, Alagille syndrome is an autosomal dominant disorder characterised by multisystem involvement. The most characteristic findings include chronic cholestasis due to intrahepatic bile duct paucity and cardiac, ocular or skeletal features with typical facies.1 Five major features described by Alagille et al. include peculiar facies, chronic cholestasis, posterior embryotoxon, butterfly-like arch defect and peripheral pulmonary artery hypoplasia.2 The incidence of Alagille syndrome has been proposed to be around 1 in 30 000.3
THE CASE
A toddler was brought to the outpatient department of our centre by the parents. They noticed poor vision in both eyes for the past 6 months and also difficulty in opening the eyes in a bright light setting, as well as keeping objects close to the face. The child was born at full term by normal vaginal delivery. There was a history of recurrent jaundice in the initial 3 months of life, for which a thorough systemic work up had been done. A heterozygous missense mutation in exon 23 of the JAG 1 gene (p. Thr962Ala) corresponding to Alagille syndrome was detected in the patient and his mother. The mother had pigmented stones in the gall bladder and had a laparoscopic cholecystectomy in the third decade of her life.
The child was also diagnosed with autism, developmental delay, hypothyroidism, and an atrial septal defect which reduced in size over time. The child had typical facies with frontal bossing, broad nasal bridge, epicanthus palpebralis and moderate hypertelorism (Fig. 1a). Lid abnormalities gave an appearance of pseudoesotropia. X-ray chest revealed butterfly-shaped vertebrae (Fig. 1b). Ophthalmic examination revealed a lamellar cataract with riders in both eyes with a good central glow. Retinoscopy (under homatropine 2%) revealed a spherical equivalent of minus 5.5 D in the right eye and minus 6 D in the left eye. A binocular visual acuity of 6/96 was noted with Cardiff cards. Glasses were prescribed and the child was on kept on 6-monthly follow-up. Because of worsening visual axis opacification and falling visual acuity on follow-up (binocular Cardiff visual acuity of 6/120), surgical intervention was planned for both the eyes. The child was admitted for examination under anaesthesia for biometry followed by surgery.
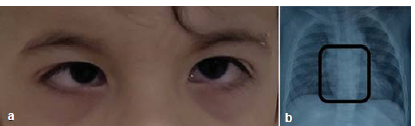
- (a) Typical facies with broad nasal bridge, epicanthus palpebralis and pseudoesotropia, (b) X-ray chest showing butterfly shaped vertebrae
A thorough ophthalmic evaluation was done under general anaesthesia. Corneal diameters were 9 and 10 mm for both the eyes vertically and horizontally, respectively. Amplitude scan (Vumax HD ophthalmic ultrasound) revealed an axial length of 25.31 mm and 25.51 mm in the right and left eye, respectively.
Keratometry (Righton Retinomax K Plus 5 handheld autorefractor/keratometer) was measured as 38.12 @ 390/39.25 D @ 1290 in the right eye and 36.12 @ 660 /39.87 @ 1560 in the left eye. The presence of pannus and posterior embryotoxon was noted (Fig. 2). The lens showed a lamellar cataract with riders along with a posterior subcapsular component (Fig. 2). Figure 3 shows ultrasound biomicroscopy (Vumax HD ophthalmic ultrasound) of both eyes that revealed a lamellar cataract with few opacities in the posterior subcapsular region.
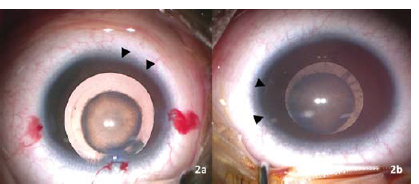
- Image of the (a) right eye showing zonular cataract with pannus (arrow heads) and (b) left eye showing zonular cataract with posterior subcapsular component with posterior embryotoxon (Arrow heads)

- Ultrasound biomicroscopy showing zonular cataract in both the eyes
Lens aspiration with posterior curvilinear capsulorhexis with anterior vitrectomy and placement of the intraocular lens in the capsular bag was done. The emmetropic IOL power for both eyes was similar (21.10 D). A hydrophobic, single-piece, foldable, acrylic, intraocular lens with a power of +20.5D (2.8% under correction), was placed in the bag in both the eyes. IOL was inserted through a superior 2.8 mm entry, which was sutured with 10-0 monofilament nylon. Intraoperative fundus examination revealed myopic fundus, peripapillary depigmentation and mild temporal disc pallor (Fig. 4). The child was prescribed moxifloxacin 0.5% four times a day, predacetate six times a day with weekly tapering over 6 weeks and homatropine four times a day for 8 weeks.
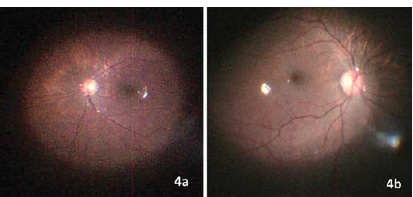
- Intraoperative retinal imaging showing myopic fundus, peripapillary depigmentation and mild temporal disc pallor in both the eyes.
Postoperatively, the patient had a good red reflex. Postoperative day 1 retinoscopy was plus 2.75 D (vertical) and 3.25 D (horizontal) in the right eye and 3.5 D in both axes in the left eye. The child was prescribed plus 3 D spherical in the right eye and 3.5 D spherical in the left eye. Postoperative Cardiff revealed a binocular vision of 6/24. Examination under anaesthesia was done at a 4-week follow-up. Sutures were removed and examination revealed a well-centred IOL with a clear visual axis in both eyes.
DISCUSSION
Alagille syndrome is caused by a mutation in the NOTCH pathway that has a role in deciding the fate of a cell during division. The NOTCH pathway is an intercellular signalling system that is essential for proper embryonic development. NOTCH 1–4 is a family of single-pass receptors that are activated by a five-membered family of transmembrane proteins including DLL1, DLL3, DLL 4, Jagged 1 and Jagged 2. Various mutations in Jagged 1 (in 94% of all patients reported to date) and NOTCH 2 (rare) have been associated with the occurrence of the syndrome.3,4
Ocular findings include the almost universal occurrence of posterior embryotoxon. Other reported associations include strabismus, microcornea, nanophthalmos, keratoconus, high myopia, iris hypoplasia, axenfeld anomaly and a few reports of cataracts. Anomalous optic disc, peripapillary depigmentation, speckling of the retinal pigment epithelium and chorioretinal atrophy have also been described.5–8
A posterior subcapsular cataract is the most common cataract morphology described in the literature in patients with Alagille syndrome. It has been described in a 28-year-old man by Riely et al. and a 31-year-old man by Puklin et al.5,7 Fukumoto described a 15-year-old girl with a posterior subcapsular cataract, who was operated at the age of 15 years and developed subluxation of the intraocular lens–capsular bag complex 4 years after surgery.9 Hingorani et al. in a case series of 22 patients described lenticular opacities in 2 patients (diffuse lenticular haziness in one and sutural opacities in the other) and cortical cataracts in parents of 8 patients.6 None of the papers reports the morphology seen in our patient.
A lamellar cataract is a genetically heterogenous entity and has been mapped to various genetic loci (Chr 1, 2, 11, 16, 17, 21). We assume that the occurrence of lamellar cataract in this child could either be a chance association or a manifestation of a larger chromosomal abnormality involving adjacent genetic loci. The novelty in our case report is a previously unreported morphology of cataracts and presentation in the initial few years of life.
To the best of our knowledge, this is the first case describing a lamellar cataract with riders in a toddler. The patient also showed other typical ocular features of Alagille syndrome including corneal pannus, posterior embryotoxon, axial myopia and chorioretinal degenerations. Systemic features included peculiar facies (prolonged head, mild hypertelorism and saddle-like nose), butterfly-shaped vertebra on thoracic X-ray, atrial septal defect, and chronic cholestasis. JAG 1 gene is located on chromosome 20p12. A larger deletion of chromosome 20p12 has been associated with the presence of additional abnormalities such as autism and developmental delay as seen in our patient.10
References
- Hepatic ductular hypoplasia associated with characteristic facies, vertebral malformations, retarded physical, mental, and sexual development, and cardiac murmur. J Pediatr. 1975;86:63-71.
- [CrossRef] [PubMed] [Google Scholar]
- Syndromic paucity of interlobular bile ducts (Alagille syndrome or arteriohepatic dysplasia): Review of 80 cases. J Pediatr. 1987;110:195-200.
- [CrossRef] [PubMed] [Google Scholar]
- NOTCH2 mutations in Alagille syndrome. J Med Genet. 2012;49:138-44.
- [CrossRef] [PubMed] [Google Scholar]
- Anterior segment and retinal pigmentary abnormalities in arteriohepatic dysplasia. Ophthalmology. 1981;88:337-47.
- [CrossRef] [PubMed] [Google Scholar]
- Ocular abnormalities in Alagille syndrome. Ophthalmology. 1999;106:330-7.
- [CrossRef] [PubMed] [Google Scholar]
- Arteriohepatic dysplasia: A benign syndrome of intrahepatic cholestasis with multiple organ involvement. Ann Intern Med. 1979;91:520-7.
- [CrossRef] [PubMed] [Google Scholar]
- Characterization of the spectrum of ophthalmic changes in patients with Alagille syndrome. Invest Ophthalmol Vis Sci. 2021;62:27.
- [CrossRef] [PubMed] [Google Scholar]
- A case of Alagille syndrome complicated by intraocular lens subluxation and rhegmatogenous retinal detachment. Clin Ophthalmol. 2013;7:1463-5.
- [CrossRef] [PubMed] [Google Scholar]
- SNP array mapping of chromosome 20p deletions: Genotypes, phenotypes, and copy number variation. Hum Mutat. 2009;30:371-8.
- [CrossRef] [PubMed] [Google Scholar]



