Translate this page into:
Neuropathogenesis by Chandipura virus: An acute encephalitis syndrome in India
Corresponding Author:
Anirban Basu
National Brain Research Centre, Manesar 122051, Haryana
India
anirban@nbrc.ac.in
| How to cite this article: Ghosh S, Basu A. Neuropathogenesis by Chandipura virus: An acute encephalitis syndrome in India. Natl Med J India 2017;30:21-25 |
Abstract
Chandipura virus (CHPV) has been contributing to the rising number of premature deaths due to acute encephalitis syndrome for over a decade in India. CHPV belongs to the family Rhabdoviridae. Neuropathogenesis of CHPV has been well established but the exact route of entry into the central nervous system (CNS) and the triggering factor for neuronal death are still unknown. Rabies virus and vesicular stomatitis virus, which are related closely to CHPV, enter the CNS retrogradely from peripheral or olfactory neurons. Disruption of the blood–brain barrier has also been connoted in the entry of CHPV into the CNS. CHPV upon entering the neurons triggers cellular stress factors and release of reactive oxygen species (ROS). The stress granules produced in response to cellular stress have been implicated in viral replication and ROS generation, which stimulates neuronal death. Both these phenomena cohesively explain the neuropathogenesis and neurodegeneration following CHPV infection.
Introduction
Acute encephalitis syndrome (AES) accounts for a majority of premature deaths in India. In most cases, it is considered to be due to bacterial meningitis or Japanese encephalitis virus (JEV) infection. However, in over 50% of patients with AES, the source or causal agent goes unrecognized.[1] Since 2000, the epidemics caused due to viral attacks in India are mainly connected to Chandipura virus (CHPV),[2] Nipah virus[3] and enteroviruses.[4],[5] Among these viruses, CHPV had the highest reported case fatality rate. With increase in travel and globalization, these viruses are no longer restricted to national boundaries. The endemic zones of CHPV in India are shown in [Figure - 1].
![[Figure - 1]](#fig_NatlMedJIndia_2017_30_1_21_210150_f1.jpg){kind=link}
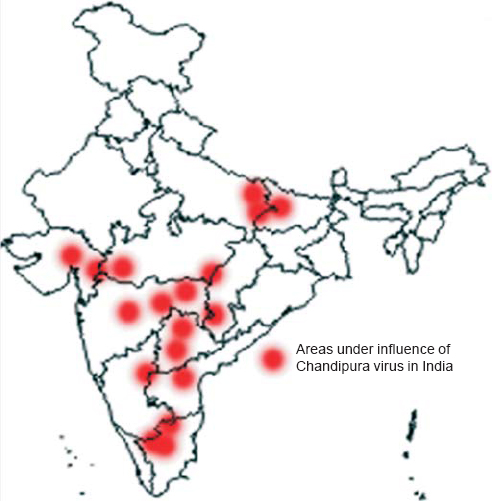 |
| Figure 1: Endemic zones of Chandipura virus in India. The dots represent the endemic regions in India affected by Chandipura virus. The map of India was adapted from the Geography Blog (http://the-geography.blogspot.in/2015/05/outlines-mapsindia.html). |
Neurodegeneration following CHPV infection has been well documented over the years although there have been lesser known facts that explain the cause of neuronal death.[6],[7] However, the route of neuro-invasion for CHPV has remained obscure.[8] We review the current information from related virological reports to propose a plausible explanation about the route of neuro-invasion and the causal agent of neurodegeneration.
Virology
CHPV belongs to the family Rhabdoviridae in the order Mononegavirales of the genus Vesiculovirus. The incessant mutating nature of CHPV has enhanced its lethal nature to cause human infections unlike its genetic cousin, the vesicular stomatitis virus (VSV). Children below 15 years of age are more vulnerable than adults who show symptoms similar to any encephalitis such as high-grade fever, vomiting, altered sensorium, generalized convulsions, decerebrate posture and grade IV coma.[9],[10]
Comparative sequence analysis concludes that CHPV is evolutionarily equidistant from new world vesiculoviruses VSV Indiana (VSVind) and VSV New Jersey (VSVnj), and closely related to its Asian kin Isfahan.[11] CHPV, like VSV, has an outer lipoprotein envelope that encloses a helical ribonucleoparticle (RNP), which further enwraps a non-segmented single-stranded RNA genome. VSV has five structural proteins, namely glycoprotein (G), matrix protein (M), large protein (L), nucleocapsid protein (N) and phosphoprotein (P) [Figure - 2]. The G protein is interspersed within the lipid bi-layer and protrudes from the outer membrane, and plays a dual role in receptor recognition and fusion of viral and cellular membranes at a low pH.[12] The M protein acts as an adhesive layer between the inner membrane of the lipid bi-layer and the core nucleocapsid.[13] The RNP core consists of two parts: (i) the core N protein that enwraps the viral genome and forms the template for the viral transcription, and (ii) viral RNA-dependent RNA polymerase (RdRp) consisting of the L protein that acts as the catalytic subunit and the P protein acting as the transcriptional activator.[14] The VSV genome is approximately 11 kb in size with a 49 nt (nucleotide) leader gene (l) followed by five transcriptional units coding for viral proteins separated by intragenic spacer regions and a short non-transcribed 46 nt trailer sequence (t) arranged in the order 3'-l-N-P-MG-L-t 5'. The life cycle of CHPV within its host is shown in [Figure - 3].
![[Figure - 2]](#fig_NatlMedJIndia_2017_30_1_21_210150_f2.jpg){kind=link}
![[Figure - 3]](#fig_NatlMedJIndia_2017_30_1_21_210150_f3.jpg){kind=link}
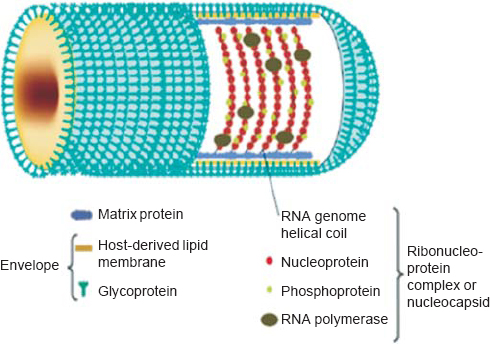 |
| Figure 2: Structure of Chandipura virus. Chandipura virus has a rodshaped structure, which is characteristic of the family Rhabdoviridae. The outer envelope consists of glycoprotein (G) and host-derived lipid membrane. The viral RNA and envelope are connected through the matrix protein (M). The genome is a single-stranded RNA having a negative helical turn. The RNA genome is associated with three other viral proteins known as nucleoprotein (N), phosphoprotein (P) and large protein (L) forming the ribonucleoprotein complex. Reprinted with permission from: Li J, Zhang Y. Messenger RNA cap methylation in vesicular stomatitis virus, a prototype of non segmented negative sense RNA virus. In: Dricu A (ed). Methylation—From DNA, RNA and histones to diseases and treatment. In Tech; 2012. (© 2012 Li J, Zhang Y. Published in [short citation] under CC BY 3.0 license. Available at http://dx.doi.org/10.5772/54598.) |
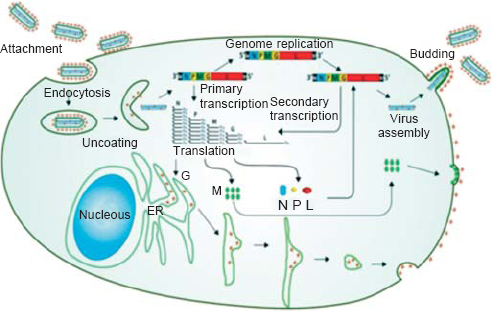 |
| Figure 3: Schematic diagram showing the life cycle of Chandipura virus within the host. Chandipura virus G protein binds with a host receptor and gets internalized through endocytic vesicles.With the lowering of pH within the vesicles, the ribonucleoprotein gets released from the envelope to enter the cytoplasm and primary transcription of the viral mRNAs begin. Viral mRNAs are transcribed in a decreasing order of molar ratio as N>P>M>G>L and enter the translation phase. The newly synthesized G protein migrates to the endoplasmic reticulum where it is packaged and secreted out in the host plasma membrane (outer) by the Golgi complex. While the translated N, P and L take part in genomic replication (to produce more viral RNA) and secondary transciption (to produce more viral mRNAs), the P protein acts as a switch that decides between replication and transcription. The translated M protein like the G protein migrates to the plasma membrane but to the inner side. The newly synthesized viral RNA is enwrapped by the N protein along with L and P and gets assembled at the inner plasma membrane near the M protein. The ribonucleoprotein complex gets bound to the M protein which further includes the host plasma membrane to form the lipid envelope. At this point when the virus gets ready to be released out of the cell, the G protein gets attached to the lipid envelope and completes the formation of a matured virion. Reprinted with permission from: Li J, Zhang Y. Messenger RNA cap methylation in vesicular stomatitis virus, a prototype of nonsegmented negative sense RNA virus. In: Dricu A (ed). Methylation—From DNA, RNA and histones to diseases and treatment. InTech; 2012. (© 2012 Li J, Zhang Y. Published in [short citation] under CC BY 3.0 license. Available at http://dx.doi.org/10.5772/54598.) |
Transmission
The vectorial capacity of mosquitoes to transmit CHPV was proved in 1967 by scientists at the National Institute of Virology, Pune.[15] A patient admitted at Vellore in 1988, who developed symptoms of encephalitis had CHPV isolated from his cerebrospinal fluid (CSF), and was suspected to have been infected by a mosquito bite, most likely Aedes aegypti.[16] Although previous reports considered mosquitoes to be potential vectors for CHPV, recent reports have suggested that sand flies play this role (Phlebotomus papatasi, Phlebotomus argentipes or Sergentomyia spp.).[17],[18] While investigating the potential vector for CHPV infection in laboratory conditions, it was found that P. argentipes not only had a high susceptibility towards CHPV infection through the oral route but was also present in areas endemic for CHPV.[19] A recent report investigated the vectorial capacity of Culex gelidus, a widely prevalent mosquito in India and Southeast Asian countries, in a range of tropical viruses including CHPV. It was observed that C. gelidus can harbour more than 5 log10TCID50 per ml of CHPV and has been postulated to be a potential vector for CHPV.[20] This transmission is of importance in investigating the life cycle of CHPV.
Neuro-Invasion Of CHPV
Zoonotic infection refers to an incursion of a virus from its natural host into a ‘dead-end host’ that has little chance to further disseminate the infection. Acute infection of the central nervous system (CNS) is an example of zoonotic infection that has apparently no selective advantage for the host or the pathogen.
Neurotropic viruses that generally have minimal pathogenic effects in their natural hosts often cause catastrophe upon entering the CNS. As mentioned earlier, CHPV enters the host system through bites of mosquito or sand fly. The host cells of the peripheral system get alarmed by pattern recognition receptors (PRRs) in response to which the innate immune system gets activated. A series of molecular events follow, which involve the release of cytokines initially, and antiviral proteins such as type I interferon's (IFN-α/β). If these fail to clear the virus, cytokines and infected cells activate the sentinel cells (e.g. dendritic cells, macrophages), which produce large amounts of cytokines and present antigens to trigger T cell-mediated immunity. The rapid innate immune response also involves activation of the complement system, natural killer (NK) cells, neutrophils and other granulocytes. Over-reaction of this response is termed as ‘cytokine storm'. This becomes fatal in case of the brain since neurons are irreplaceable and hence the infected neurons cannot be easily eliminated by canonical T cell-mediated cyto lysis. The strategy involves release of virus-specific antibodies and interferon gamma (IFNγ) from T cells.[21]
The CNS is a well-protected organ of the mammalian system, isolated from the peripheral system and guarded by the blood–brain barrier (BBB), which restricts entry of most large molecules and pathogens circulating in the blood. Neurotropic viruses such as CHPV are opportunistic pathogens that find their way into the brain escaping the immune system of the peripheral organs. Although the exact route of CHPV entry into the CNS is still unknown, various hypotheses based on research of VSV and rabies virus (RV) can be postulated to trace the footsteps of CHPV into the CNS.
Chpv Entry Into The CNS
When suckling Balb/c mouse pups were injected with CHPV through their footpads, viral replication increased in the blood and other peripheral organs (e.g. thymus, spleen, liver, kidney, heart, lungs) till 24 hours post-infection and then decreased. However, in the spinal cord and brain viral replication continued to show an increasing in the trend. After 24 hours of infection, detectable amounts of viral RNA were seen only in the spinal cord (both lower and upper) but not in the cerebrum. After 96 hours of infection, viral titres increased in all neuronal tissues except the sciatic nerves.[8] However, these results do not explain the route of migration of CHPV to the CNS but establish the neurotropism of this virus.
CHPV and VSV enter the cell through endocytosis after binding to a host receptor [Figure - 3]. In case of VSVind, it has been reported that the G protein binds to LDL receptor while RV binds to neuron cell adhesion molecules (NCAM) and nicotinic acetylcholine receptors (nAchR).[22] It may be hypothesized that on entering the host through the bloodstream, CHPV infects various peripheral organs and quickly migrates towards the peripheral nerve terminals that harbour several cellular receptors. RV has been observed to enter the CNS through olfactory neurons, thus the possibility of CHPV entering the CNS through this route should also be taken into consideration.[23] Upon binding to the specific receptors as mentioned above, CHPV gets endocytosed through a clathrin-mediated mechanism into special endocytic vesicles where a neutral pH is maintained till the point when the virus requires to move into the host cytoplasm.
The virus packed inside the endocytic vesicle upon entering the axonal terminal of the neurons binds to the (−) end-directed dynein motors for retrograde transport on microtubules towards the cell body.[24] Some studies have proposed the role of the viral P protein in mediating the retrograde transport with the help of dynein, though this theory was later rebutted.[25] Other hypotheses state that neurotrophins and brain-derived neurotrophic growth factor (BDNF) and their receptors (e.g. p75NTR and TrkB) get incorporated into Rab5-positive early endosome-like vesicles, which mature into Rab7-positive late endosome-like vesicles. Later, Rab7 mediates the recruitment of the dynactin (p50/glued) part of the dynein complex, which further undergoes retrograde transport to the cell body.[26] Although these theories provide some insight into the molecular mechanisms of post-entry retrograde transport, it is not clear whether virions endocytosed at the nerve terminal get engaged in a default retrograde transport pathway.
It was shown earlier that CHPV infection damages the BBB.[9] Hence, the possibility of CHPV breaching the BBB and entering the CNS cannot be negated. Pericytes, astrocyte end feet projections, brain microvascular endothelial cells (BVMECs), basement membrane cell and microglial cells that ensheath the capillary wall form the BBB. It has been reported that the BBB can be breached in viral infections by either the virus passing through it or infecting the BBB cells and entering the CNS.[27]
Post-Replication Transport, Egress And Spread Of CHPV
Biosynthetic machinery (e.g. ribosomes and secretory pathway organelles) are distributed in axons, dendrites and the cell body of neurons, which are used by the virus to support their survival within the cell.[28] However, no study has conclusively defined whether this distribution is used for viral replication and gene expression in a compartment-specific manner. Thus, it is difficult to state at which point the endocytic vesicles lower pH to facilitate the release of the viral RNP into the cell cytoplasm and start viral replication. Post-replication the virus again requires the microtubule motors to transport their viral components and sub-assemblies.[29] It has been observed that RV G proteins spread across synaptic connections in a retrograde manner, while VSV G proteins follow an antegrade mode of transport.[30]
Enveloped viruses such as CHPV, VSV and RV exit cells by budding through cellular membranes of the host to derive the lipid bi-layer envelope.[31] This process enhances the virulence facilitating these viruses to infect more cells in contact with the infected host cell. It has been observed that virions do not spread by free diffusion within the CNS, but transmit between tightly connected neurons via neurochemical synapses or other cell-to-cell interconnections.[32] Hence, the spread of these viruses maintains a certain polarity in direction when moving up through the olfactory neurons or spinal cord towards the brain.
Immunological Response Against CHPV Infection
Mitogen stimulated peripheral blood mononuclear cells (PBMC) supernatants collected from patients of the 2003 outbreak in Andhra Pradesh were analysed for cytokine levels that showed significant increase in IL-2, IFN-γ, TNF-α and IL-6.[33] CHPV infection in mouse model showed significant rise in TNFα, IFNγ, MCP-1, IL-6, IL-10 levels in 24 hours post-infection except IL- 12p70 that was observed to increase at 72 hours post-infection, while others showed a decrease in trend over the course of infection.[9] The early rise in the levels of cytokines and chemokines can be correlated to the virus entering the peripheral system and this may explain the breach in the BBB in the early phase of infection. With the clearance of the virus from the peripheral system, which has been correlated with the increase in IgM at 24 hours post-infection,[9] the levels of cytokines and chemokines also reduced. However, the late activation of IL-12p70 remains controversial.
CHPV upon entering the CNS has been reported to induce microglial activation characterized by the release of IL-6, MCP- 1, iNOS and COX-2, specifically in the cortical and hippocampal regions of the brain.[34] This report suggests that the region-specific microglial activation stimulates the bystander killing of neurons following CHPV infection. Hence, the age-old controversy regarding whether CHPV infection results into encephalitis, encephalopathy or acute brain attack gets a realistic answer.
Endoplasmic Reticulum and Oxidative Stress: Hallmarks of Viral Infection
Reactive oxygen species (ROS) is a double-edged sword in terms of cellular homeostasis. ROS is generated in high levels in response to any external attack to the cell or any kind of pathogenic attack or disease conditions including cancer, diabetes and neurodegenerative diseases.[35],[36] In cases of pathogenic attacks, ROS is generated to stimulate cytokines such as TNFa, IL-6, which further trigger the adaptive immune response.[37] On the other hand, ROS production is an early signal for cells undergoing apoptosis. A very delicate balance exists between uncontrolled ROS generation and ROS generation as a mode of cellular protective response that is controlled by the feedback mechanism of secretion of antioxidant species.
Under physiological conditions, ROS are generated as byproducts of the mitochondrial respiratory chain, arachidonic acid pathway, cytochrome P450 family, glucose oxidase, amino acid oxidases, xanthine oxidase, NADPH/NADPH oxidases or NO synthases.[38] As mentioned earlier, ROS accumulation alarms various endogenous antioxidant defence systems that include both enzymatic and non-enzymatic antioxidant mechanisms, which can either scavenge ROS or prevent their formation. The enzymatic antioxidant defence mechanisms are modulated through superoxide dismutase (SOD), glutathione peroxidase (GPX), catalase and thioredoxin reductase.[39] Anti-oxidants may also be obtained through dietary intake as for example vitamins provide a non-enzymatic antioxidant defence.[40] Redox homeostasis is also contributed by NAD/NADH, NADP/NADPH and oxidized glutathione/reduced glutathione (GSSG/GSH) systems.[41]
RNA viruses on entering the cell try to hack the host metabolic machinery and in the process interfere with the cell's ROS system.[38] ROS generation mediates the leakage of Ca2+ from the lumen of the ER on being stimulated through the ER or oxidative stress.[42] This increase in Ca2+ stimulates generation of mitochondrial ROS through multiple pathways, as mentioned earlier. Hence, the mitochondrial electron transport chain generates ROS as a result of accumulation of mitochondrial Ca2+. The amount of mitochondrial ROS production is wired with the quantity of the ubisemiquinone radical intermediate (QH), an intermediate in the Q cycle at complex III.[43] The level of QH increases inversely to the activation of complex III. Ca2+ infiltration in mitochondria creates pores in the inner mitochondrial membrane that helps in leakage of cytochrome c, thereby blocking the respiratory chain at complex III. Simultaneously, the generation of QH is increased when the respiratory chain turns over more quickly. Ca2+ leak again stimulates the TCA cycle, thus increasing O2 consumption and ROS generation.[44] Other reports have suggested that Ca2+ also induces the production of nitric oxide synthase, which generates NO that inhibits complex IV and can thereby enhance ROS production.[45],[46]
Since both ER and mitochondria are in close proximity to each other, Ca2+ release from the mitochondrial matrix also infiltrates the ER membrane and enhances ER stress finally resulting in apoptosis.[38]
Thus, ER[47] and oxidative stress[48] are the two hallmarks of viral infection in the cell that lead to apoptosis mediated by several intermediate molecules on activation by dysregulated ROS generation. Several reports over the years have associated the process of viral replication and ROS production in cells but the exact mechanism is still elusive.[38] One report on VSV postulates that it induces the production of stress granule (SG)-like structures that are usually produced in response to heat, shock or oxidative stress, within which they replicate.[49] These SG-unlike granules released in response to oxidative stress do not have the usual markers such as eukaryotic initiation factor 3 (eIF3) or eIF4A, or processing body (PB) markers such as mRNA-decapping enzyme 1A (DCP1a). Furthermore, VSV has been found not to interfere with the formation of physiological SG. While some viruses have been reported to induce the formation of such SGs in their earlier phases of infection, other viruses inhibit their formation, proposing that these SGs have an antiviral response to infection.[50],[51]
Conclusion
Deciphering the route of virus entry into the brain and thereafter the neurons is important in the context of neurotropic virus infections. CHPV enters the CNS by moving retrogradely from either peripheral or olfactory neurons. Other theories suggest that the release of large amounts of cytokines and chemokines in response to peripheral infection by CHPV helps in degrading the BBB, which assists CHPV to breach through the defences of the CNS. It is interesting to note that cellular stress factors are not only secreted as a host response to viral infection but also play an important role in viral propagation within the host. Present studies indicate that CHPV either induces neuronal death directly by triggering the death domains[6] or through a bystander mechanism involving microglial activation[34] although the triggering factor for the initiation of death is not well established. Through this review we have tried to connect the two phenomena: viral entry into the CNS and the role played by stress factors initiated by viral entry into their target hosts, i.e. neurons,[7] to describe the neuropathogenesis of CHPV infection leading to neurodegeneration.
Acknowledgements
The study was supported by the core funds of the National Brain Research Centre and a grant from the Department of Biotechnology (BT/PR7907/ MED/29/702/2013), Government of India, to AB. AB is also a recipient of the Tata Innovation Fellowship (BT/HRD/35/01/02/2014) from the Department of Biotechnology, Government of India.
Conflict of interest. None declared
| 1. | Kelly R. Acute encephalitis syndrome outbreaks in India—an ongoing puzzle. Available at https://sphcm. med. unsw. edu. au/infectious-diseases-blog/acute-encephalitis-syndrome-outbreaks-india-%E2%80%93-ongoing-puzzle (accessed on 15 Jun 2016). [Google Scholar] |
| 2. | Rao BL, Basu A, Wairagkar NS, Gore MM, Arankalle VA, Thakare JP, et al. A large outbreak of acute encephalitis with high fatality rate in children in Andhra Pradesh, India, in 2003, associated with Chandipura virus. Lancet 2004;364:869–74. [Google Scholar] |
| 3. | Chadha MS, Comer JA, Lowe L, Rota PA, Rollin PE, Bellini WJ, et al. Nipah virus- associated encephalitis outbreak, Siliguri, India. Emerg Infect Dis 2006;12:235–0. [Google Scholar] |
| 4. | Joshi R, Kalantri SP, Reingold A, Colford JM Jr. Changing landscape of acute encephalitis syndrome in India: A systematic review. Natl Med J India 2012;25:212–20. [Google Scholar] |
| 5. | George S, Yergolkar PN, Kamala H, Kamala CS. Outbreak of encephalitis in Bellary District of Karnataka and adjoining areas of Andhra Pradesh. Indian J Med Res 1990;91:328–30. [Google Scholar] |
| 6. | Ghosh S, Dutta K, Basu A. Chandipura virus induces neuronal death through Fas- mediated extrinsic apoptotic pathway. J Virol 2013;87:12398–406. [Google Scholar] |
| 7. | Ghosh S, Mukherjee S, Basu A. Chandipura virus perturbs cholesterol homeostasis leading to neuronal apoptosis. J Neurochem 2015;135:368–80. [Google Scholar] |
| 8. | Anukumar B, Amirthalingam BG, Shelke VN, Gunjikar R, Shewale P. Neuro-invasion of Chandipura virus mediates pathogenesis in experimentally infdected mice. Int J Clin Exp Pathol 2013;6:1272–81. [Google Scholar] |
| 9. | Balakrishnan A, Mishra AC. Immune response during acute Chandipura viral infection in experimentally infected susceptible mice. Virol J 2008;5:121. [Google Scholar] |
| 10. | Gurav YK, Tandale BV, Jadi RS, Gunjikar RS, Tikute SS, Jamgaonkar AV, et al. Chandipura virus encephalitis outbreak among children in Nagpur division, Maharashtra, 2007. Indian J Med Res 2010;132:395–9. [Google Scholar] |
| 11. | Marriott AC. Complete genome sequences of Chandipura and Isfahan vesiculoviruses. Arch Virol 2005;150:671–80. [Google Scholar] |
| 12. | Baquero E, Albertini AA, Raux H, Buonocore L, Rose JK, Bressanelli S, et al. Structure of the low pH conformation of Chandipura virus G reveals important features in the evolution of the vesiculovirus glycoprotein. PLoS Pathog 2015;11:e1004756. [Google Scholar] |
| 13. | Ahmed M, McKenzie MO, Puckett S, Hojnacki M, Poliquin L, Lyles DS. Ability of the matrix protein of vesicular stomatitis virus to suppress beta interferon gene expression is genetically correlated with the inhibition of host RNA and protein synthesis. J Virol 2003;77:4646–57. [Google Scholar] |
| 14. | Basak S, Mondal A, Polley S, Mukhopadhyay S, Chattopadhyay D. Reviewing Chandipura: A vesiculovirus in human epidemics. Biosci Rep 2007;27:275–98. [Google Scholar] |
| 15. | Mavale MS, Geevarghese G, Ghodke YS, Fulmali PV, Singh A, Mishra AC. Vertical and venereal transmission of Chandipura virus (Rhabdoviridae) by Aedes aegypti (Diptera: Culicidae). J Med Entomol 2005;42:909–11. [Google Scholar] |
| 16. | Menghani S, Chikhale R, Raval A, Wadibhasme P, Khedekar P. Chandipura virus: An emerging tropical pathogen. Acta Trop 2012;124:1–14. [Google Scholar] |
| 17. | Mavale MS, Fulmali PV, Geevarghese G, Arankalle VA, Ghodke YS, Kanojia PC, et al. Venereal transmission of Chandipura virus by Phlebotomus papatasi (Scopoli). Am J Trop Med Hyg 2006;75:1151–2. [Google Scholar] |
| 18. | Fontenille D, Traore-Lamizana M, Trouillet J, Leclerc A, Mondo M, Ba Y, et al. First isolations of arboviruses from phlebotomine sand flies in West Africa. Am J Trop Med Hyg 1994;50:570–4. [Google Scholar] |
| 19. | Mavale MS, Fulmali PV, Ghodke YS, Mishra AC, Kanojia P, Geevarghese G. Experimental transmission of Chandipura virus by Phlebotomus argentipes (diptera: psychodidae). Am J Trop Med Hyg 2007;76:307–9. [Google Scholar] |
| 20. | Sudeep AB, Ghodke YS, George RP, Ingale VS, Dhaigude SD, Gokhale MD. Vectorial capacity of Culex gelidus (Theobald) mosquitoes to certain viruses of public health importance in India. J Vector Borne Dis 2015;52:153–8. [Google Scholar] |
| 21. | Griffin DE, Metcalf T. Clearance of virus infection from the CNS. Curr Opin Virol 2011;1:216–21. [Google Scholar] |
| 22. | Ugolini G. Advances in viral transneuronal tracing. J Neurosci Methods 2010;194: 2–20. [Google Scholar] |
| 23. | Mori I, Goshima F, Ito H, Koide N, Yoshida T, Yokochi T, et al. The vomeronasal chemosensory system as a route of neuroinvasion by herpes simplex virus. Virology 2005;334:51–8. [Google Scholar] |
| 24. | Beier KT, Saunders AB, Oldenburg IA, Sabatini BL, Cepko CL. Vesicular stomatitis virus with the rabies virus glycoprotein directs retrograde transsynaptic transport among neurons in vivo. Front Neural Circuits 2013;7:11. [Google Scholar] |
| 25. | Mebatsion T. Extensive attenuation of rabies virus by simultaneously modifying the dynein light chain binding site in the P protein and replacing Arg333 in the G protein. J Virol 2001;75:11496–502. [Google Scholar] |
| 26. | Deinhardt K, Salinas S, Verastegui C, Watson R, Worth D, Hanrahan S, et al. Rab5 and Rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron 2006;52:293–305. [Google Scholar] |
| 27. | Spindler KR, Hsu TH. Viral disruption of the blood–brain barrier. Trends Microbiol 2012;20:282–90. [Google Scholar] |
| 28. | Hanus C, Ehlers MD. Secretory outposts for the local processing of membrane cargo in neuronal dendrites. Traffic 2008;9:1437–5. [Google Scholar] |
| 29. | LaVail JH, Tauscher AN, Aghaian E, Harrabi O, Sidhu SS. Axonal transport and sorting of herpes simplex virus components in a mature mouse visual system. J Virol 2003;77:6117–26. [Google Scholar] |
| 30. | Beier KT, Saunders A, Oldenburg IA, Miyamichi K, Akhtar N, Luo L, et al. Anterograde or retrograde transsynaptic labeling of CNS neurons with vesicular stomatitis virus vectors. Proc Natl Acad Sci U S A 2011;108:15414–19. [Google Scholar] |
| 31. | Lorizate M, Kräusslich HG. Role of lipids in virus replication. Cold Spring Harb Perspect Biol 2011;3:a004820. [Google Scholar] |
| 32. | Koyuncu OO, Hogue IB, Enquist LW. Virus infections in the nervous system. Cell Host Microbe 2013;13:379–93. [Google Scholar] |
| 33. | Tripathy A, Balaji S, Rao N, Thakare J, Mishra A, Arankalle V. Cytokine levels in Chandipura virus associated encephalopathy in children. Scand J Infect Dis 2005;37:590–3. [Google Scholar] |
| 34. | Verma AK, Ghosh S, Pradhan S, Basu A. Microglial activation induces neuronal death in Chandipura virus infection. Sci Rep 2016;6:22544. [Google Scholar] |
| 35. | Liou GY, Storz P. Reactive oxygen species in cancer. Free Radic Res 2010;44: 479–96. [Google Scholar] |
| 36. | Manton KG, Volovik S, Kulminski A. ROS effects on neurodegeneration in Alzheimer's disease and related disorders: On environmental stresses of ionizing radiation. Curr Alzheimer Res 2004;14:277–93. [Google Scholar] |
| 37. | Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis. Annu Rev Immunol 2009;27:165–97. [Google Scholar] |
| 38. | Reshi ML, Su YC, Hong JR. RNA Viruses: ROS-mediated cell death. Int J Cell Biol 2014;2014:467452. [Google Scholar] |
| 39. | Sinha AK, AbdElgawad H, Giblen T, Zinta G, De Rop M, Asard H, et al. Antioxidative defences are modulated differentially in three freshwater teleosts in response to ammonia-induced oxidative stress. PLoS One 2014;9:e95319. [Google Scholar] |
| 40. | Zortea K, Fernandes BS, Guimãraes LR, Francesconi LP, Lersch C, Gama CS, et al. Reduced serum non-enzymatic antioxidant defense and increased lipid peroxidation in schizophrenic patients on a hypocaloric diet. Neurosci Lett 2012;512:43–7. [Google Scholar] |
| 41. | Ghosh D, Levault KR, Brewer GJ. Relative importance of redox buffers GSH and NAD(P)H in age-related neurodegeneration and Alzheimer disease-like mouse neurons. Aging Cell 2014;13:631–40. [Google Scholar] |
| 42. | Bhandary B, Marahatta A, Kim HR, Chae HJ. An involvement of oxidative stress in endoplasmic reticulum stress and its associated diseases. Int J Mol Sci 2012;14: 434–56. [Google Scholar] |
| 43. | Eckert A, Keil U, Kressmann S, Schindowski K, Leutner S, Leutz S, et al. Effects of EGb 761 Ginkgo biloba extract on mitochondrial function and oxidative stress. Pharmacopsychiatry 2003;36 Suppl 1:S15–S23. [Google Scholar] |
| 44. | Görlach A, Klappa P, Kietzmann T. The endoplasmic reticulum: Folding, calcium homeostasis, signaling, and redox control. Antioxid Redox Signal 2006;8:1391–418. [Google Scholar] |
| 45. | Murphy MP. How mitochondria produce reactive oxygen species. Biochem J 2009;417:1–13. [Google Scholar] |
| 46. | Meares GP, Hughes KJ, Naatz A, Papa FR, Urano F, Hansen PA, et al. IRE1- dependent activation of AMPK in response to nitric oxide. Mol Cell Biol 2011;31:4286–97. [Google Scholar] |
| 47. | Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat Rev Drug Discov 2008;7:1013–30. [Google Scholar] |
| 48. | Schwarz KB. Oxidative stress during viral infection: A review. Free Radic Biol Med 1996;21:641–9. [Google Scholar] |
| 49. | Dinh PX, Beura LK, Das PB, Panda D, Das A, Pattnaik AK. Induction of stress granule-like structures in vesicular stomatitis virus-infected cells. J Virol 2013;87: –83. [Google Scholar] |
| 50. | Iseni F, Garcin D, Nishio M, Kedersha N, Anderson P, Kolakofsky D. Sendai virus trailer RNA binds TIAR, a cellular protein involved in virus-induced apoptosis. EMBO J 2002;21:5141–50. [Google Scholar] |
| 51. | White JP, Lloyd RE. Poliovirus unlinks TIA1 aggregation and mRNA stress granule formation. J Virol 2011 ;85:12442–54. [Google Scholar] |
Fulltext Views
6,628
PDF downloads
2,679



