Translate this page into:
Overlapping polyclonal lymphoproliferative disorders
2 IRCAD, Interdisciplinary Research Center of Autoimmune Diseases, Novara, Italy
3 Department of Translational Medicine, Università del Piemonte Orientale UPO, Novara, Italy
4 Department of Pathology, Università del Piemonte Orientale UPO, Novara, Italy
Corresponding Author:
Francesco Gavelli
Università del Piemonte Orientale UPO, via Solaroli 17, 28100 Novara
Italy
gavelli.francesco@gmail.com
| How to cite this article: Bonometti R, Bellan M, Sola D, Gibbin A, Gavelli F, Patrucco F, Spina P, Gualerzi A, Favretto S, Castello LM, Sainaghi PP, Pirisi M. Overlapping polyclonal lymphoproliferative disorders. Natl Med J India 2020;33:344-346 |
Abstract
Multicentric Castleman disease (MCD) is a rare clinical entity characterized by a polyclonal lymphoid proliferation, leading to generalized lymphadenopathy, organomegaly and systemic symptoms. It has been reported in association with either other monoclonal or polyclonal lymphoid disorders, such as POEMS syndrome and immunoglobulin (Ig)G4-related disease. We present a patient showing a variant of MCD, sharing common features with POEMS syndrome and associated with the proliferation of IgG4-producing plasma cells.Introduction
Castleman disease (CD) is an uncommon lymphoproliferative disorder.[1] It is historically classified as unicentric, when a single lymph node is involved, showing the classical ‘Castleman-like’ histopathological changes, or multicentric CD (MCD);[2] a subset of MCD is caused by human herpesvirus-8 (HHV-8), whereas HHV-8-negative MCD cases remain idiopathic.[3] Anecdotal cases of overlap with other lymphoproliferative conditions have been reported in the literature; in particular, MCD has been associated with immunoglobulin (Ig)G4-related disease (IgG4-RD), a broad collection of disorders, sharing common features: tumour-like swelling of involved organs with lymphoplasmacytic infiltrate enriched in IgG4-positive plasma cells and a variable degree of fibrosis.[4],[5],[6] Moreover, MCD has been related to POEMS, a syndrome characterized by polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy and skin changes.[7]
We report a middle-aged male whose disease fulfilled the diagnostic criteria for MCD while sharing the clinical features of POEMS syndrome and IgG4-RD.
The Case
A 56-year-old male was admitted to the emergency room of a university hospital due to shortness of breath associated with anasarca. The patient complained of recent onset of bilateral lower limb pain, associated with dysaesthesia. No chest pain, fever, weight loss or pruritus was reported. His medical history was unremarkable, except for previous alcohol abuse (the patient reported complete abstinence in the past few months) and smoking (60 pack-years).
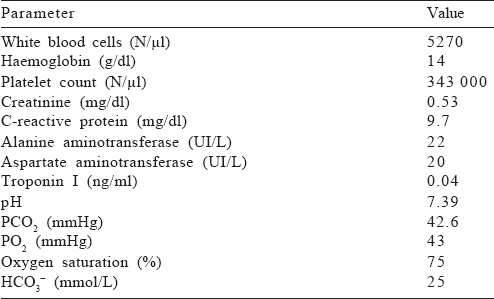
[Table - 1] gives the main laboratory findings at admission, documenting respiratory failure and elevation of inflammatory markers. An accentuation of interstitial tissue and bilateral effusion were evident on chest X-ray. Electrocardiogram revealed a sinus tachycardia.
![[Table - 1]](#tbl_NatlMedJIndia_2020_33_6_344_321144_t2.jpg){kind=link}
On clinical examination, skin hyperpigmentation and several confluent, pomphoid, non-itching skin lesions were present on the back and forearms; multiple, painless, lateral cervical and inguinal lymphadenopathy, hepatosplenomegaly and bilateral pleural effusion were noted. The patient was admitted to an internal medicine ward and underwent the following investigations:
- Echocardiogram showed a dilated cardiomyopathy with severe reduction of ejection fraction (EF; 29%) and functional mitral and tricuspid insufficiency.
- Chest and abdomen computed tomography showed bilateral lung effusion, hepatosplenomegaly and multiple mediastinal, abdominal and inguinal lymphadenopathy.
- Laboratory data showed that autoantibodies (including antinuclear, anti-extractable nuclear antigens and anti-neutrophil cytoplasmic) were negative, while a polyclonal hypergammaglobulinaemia was reported (IgG 2960 mg/dl; IgA 1050 mg/dl; IgM 138 mg/dl) with elevation of the IgG4 fraction (601 mg/dl). Moreover, adrenal insufficiency was diagnosed on the basis of a plasma cortisol concentration <1 μg/dl measured at 6 a.m.; fasting plasma glucose and HbA1c were diagnostic of new-onset diabetes mellitus, and urinalysis showed non-selective proteinuria in the nephrotic range, with preserved renal function.
- Viral and bacterial serology for Epstein–Barr virus, cytomegalovirus, HIV, HHV-8, mycobacteria and Bartonella were negative.
A periumbilical biopsy ruled out amyloid accumulation. The histological examination of a excised lymph node showed lymphoid follicles, polyclonal CD138+ plasma cells occupying interfollicular spaces; IgG4 staining was positive in a sizeable proportion of plasma cells [Figure - 1].
![[Figure - 1]](#fig_NatlMedJIndia_2020_33_6_344_321144_f1.jpg){kind=link}
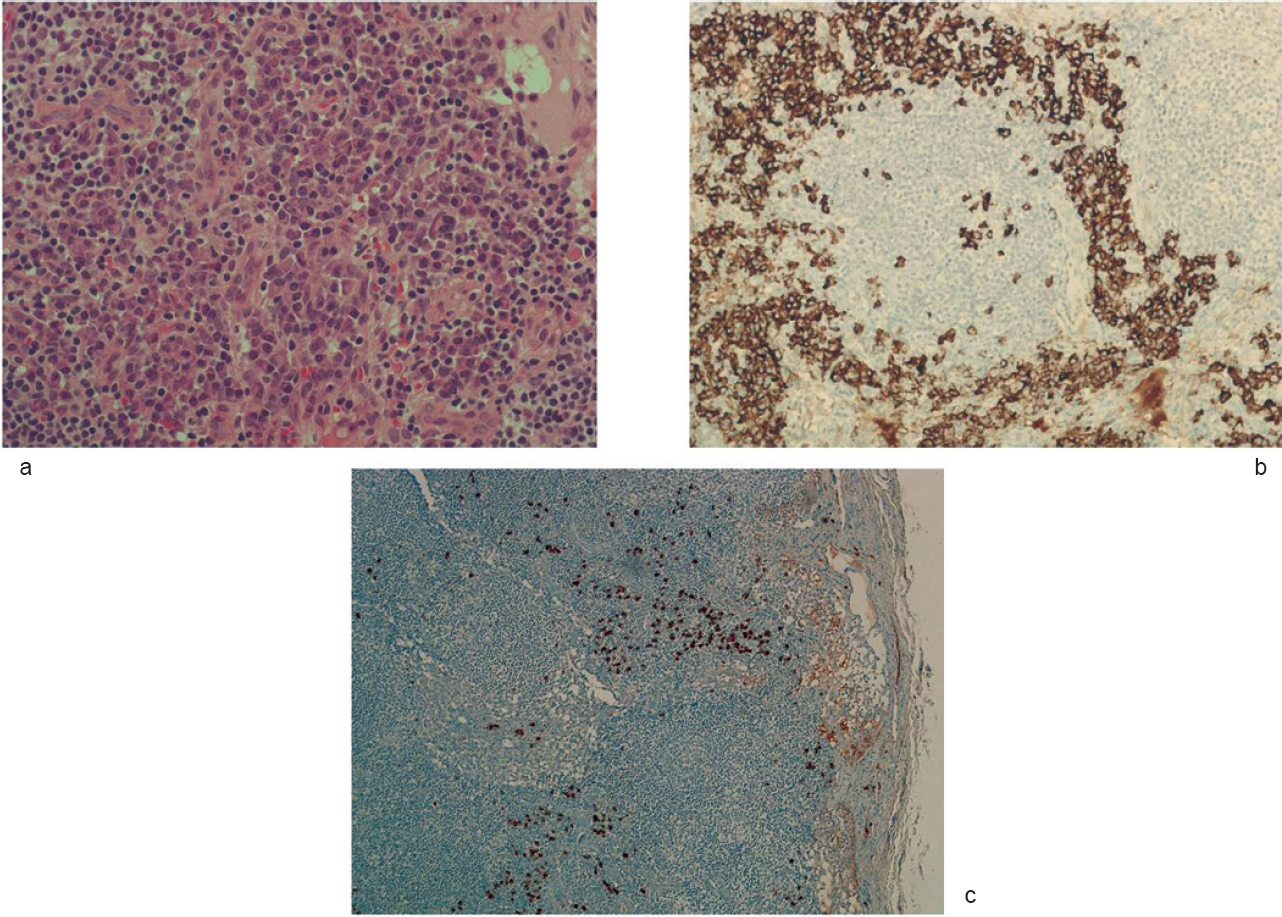 |
| Figure 1: Lymph node histopathological findings. Castleman disease, plasma cell type. Diffuse plasma cell proliferation (H and E, ×200) in the interfollicular tissue (a); underlined by immunostaining for CD138 (×200; b). Inconspicuous hyaline-vascular changes in the follicles. Immunostaining for IgG4 (×100; c) shows positive cells in the interfollicular regions |
Discussion
The clinical scenario of the patient reported here can be summarized as follows: dilated cardiomyopathy, associated with organomegaly; generalized lymphadenopathy with a polyclonal B signature and skin and endocrine manifestations.
On this basis, and according to the histopathological findings, we made a diagnosis of MCD,[8] in its idiopathic variant, being the patient negative for HHV-8. Indeed, fever, night sweats, weight loss and fatigue are common in MCD. Physical examination is typically notable for generalized lymphadenopathy and hepatosplenomegaly, and many patients have evidence of fluid retention with lower extremity oedema, pleural and pericardial effusion and abdominal ascites. Anaemia, elevated inflammatory markers, hypergammaglobulinaemia and hypoalbuminaemia are often present.[9] Our patient shared many of these clinical features; recently, an international consensus provided new diagnostic criteria for idiopathic MCD. The retrospective application of these criteria to our patient confirms the diagnosis.[3] A key aspect of this case is the concomitant presence of heart failure, which might be explained by different potential causes. First, an alcoholic cardiomyopathy is a potential explanation, following the reported alcohol abuse. However, although rare, POEMS has been shown to be associated with heart failure. In a previous cohort of POEMS patients, 3 of 99 patients presented with heart failure and cardiomyopathy that resolved following the treatment of POEMS syndrome. Four other patients developed heart failure during an exacerbation of POEMS syndrome.[10] In our patient, EF improved after treatment from 29% to 37%, suggesting a potential aetiological component from POEMS syndrome.
Our patient has overlapping features of other monoclonal and polyclonal lymphoproliferative disorders, which suggest common pathophysiological mechanisms. While MCD is present in 15%–25% of patients with POEMS,[11] our patient has some features of POEMS (organomegaly; endocrinopathy, characterized by diabetes and adrenal insufficiency; and skin changes), although a proper diagnosis cannot be made because of the absence of monoclonal gammopathy, which is a mandatory criterion.[12] It is, therefore, reasonable to consider these manifestations as the result of proliferation of B cell lineage, either monoclonal or polyclonal, rather than the effect of specific disease mechanisms. Furthermore, the proliferating plasma cells lead to a considerable increase of IgG4 fraction, usually highly specific for IgG4-RD.[13] In our patient, the lymph nodes showed proliferation of IgG4-positive plasma cells, although no other typical clinical features of IgG4-RD were evident.
Based on the clinical diagnosis, the patient was started on a regimen of steroids, with administration of a four-dose rituximab treatment, along with supportive care for his cardiac failure. The choice of rituximab was made because of the B-driven origin of the patient’s condition. CD20 targeting is considered one of the strategies of choice in MCD, despite the lack of clinical trials directly assessing the efficacy of rituximab in this context.[14] Moreover, rituximab is an effective treatment in IgG4-RD.[15] Hence, we decided to use rituximab, which induced a considerable clinical improvement, allowing discharge of the patient who started an outpatient follow-up. However, 5 months later, he had sudden cardiac death.
Conclusion
This is a clinical case of overlapping lymphoproliferative disorders. Some of the clinical entities that we use to label our patients have relatively ill-defined borders. A diagnostic approach based on pathophysiology sometimes precludes categorization into neat classifications. Our case suggests that patients classified as having polyclonal lymphoproliferative disorders such as CD when the proliferating plasma cells produce this subtype IgG4 may present with clinical features of POEMS and/or IgG4-related disorders.
Conflicts of interest. None declared
| 1. | Case records of the Massachusetts general hospital weekly clinicopathological exercises: Case 40011. N Engl J Med 1954;250:26–30. [Google Scholar] |
| 2. | Waterston A, Bower M. Fifty years of multicentric Castleman’s disease. Acta Oncol 2004;43:698–704. [Google Scholar] |
| 3. | Fajgenbaum DC, Uldrick TS, Bagg A, Frank D, Wu D, Srkalovic G, et al. International, evidence-based consensus diagnostic criteria for HHV-8-negative/ idiopathic multicentric Castleman disease. Blood 2017;129:1646–57. [Google Scholar] |
| 4. | Mochizuki H, Kato M, Higuchi T, Koyamada R, Arai S, Okada S, et al. Overlap of IgG4-related disease and multicentric Castleman’s disease in a patient with skin lesions. Intern Med 2017;56:1095–9. [Google Scholar] |
| 5. | Bellan M, Boggio E, Sola D, Gibbin A, Gualerzi A, Favretto S, et al. Association between rheumatic diseases and cancer: Results from a clinical practice cohort study. Intern Emerg Med 2017;12:621–7. [Google Scholar] |
| 6. | Izumi Y, Takeshita H, Moriwaki Y, Hisatomi K, Matsuda M, Yamashita N, et al. Multicentric Castleman disease mimicking IgG4-related disease: A case report. Mod Rheumatol 2017;27:174–7. [Google Scholar] |
| 7. | Andhavarapu S, Jiang L. POEMS syndrome and Castleman disease. Blood 2013;122:159. [Google Scholar] |
| 8. | Cheuk W, Chan JK. IgG4-related sclerosing disease: A critical appraisal of an evolving clinicopathologic entity. Adv Anat Pathol 2010;17:303–32. [Google Scholar] |
| 9. | Soumerai JD, Sohani AR, Abramson JS. Diagnosis and management of Castleman disease. Cancer Control 2014;21:266–78. [Google Scholar] |
| 10. | Dispenzieri A, Kyle RA, Lacy MQ, Rajkumar SV, Therneau TM, Larson DR, et al. POEMS syndrome: definitions and long-term outcome. Blood 2003;101: 2496–506. [Google Scholar] |
| 11. | Bélec L, Mohamed AS, Authier FJ, Hallouin MC, Soe AM, Cotigny S, et al. Human herpesvirus 8 infection in patients with POEMS syndrome-associated multicentric Castleman’s disease. Blood 1999;93:3643–53. [Google Scholar] |
| 12. | Dispenzieri A. POEMS syndrome: 2017 Update on diagnosis, risk stratification, and management. Am J Hematol 2017;92:814–29. [Google Scholar] |
| 13. | Hao M, Liu M, Fan G, Yang X, Li J. Diagnostic value of serum IgG4 for IgG4-related disease: A PRISMA-compliant systematic review and meta-analysis. Med (Baltimore) 2016;95:e3785. [Google Scholar] |
| 14. | van Rhee F, Greenway A, Stone K. Treatment of idiopathic Castleman disease. Hematol Oncol Clin North Am 2018;32:89–106. [Google Scholar] |
| 15. | Carruthers MN, Topazian MD, Khosroshahi A, Witzig TE, Wallace ZS, Hart PA, et al. Rituximab for IgG4-related disease: A prospective, open-label trial. Ann Rheum Dis 2015;74:1171–7. [Google Scholar] |
Fulltext Views
1,428
PDF downloads
409



