Translate this page into:
Venous thromboembolism due to hyperhomocysteinaemia and tuberculosis
Corresponding Author:
Jyotsna M Joshi
Department of Pulmonary Medicine, T.N. Medical College and B.Y.L. Nair Hospital, Mumbai 400008, Maharashtra
India
drjoshijm@gmail.com
| How to cite this article: Chaudhary A, Desai U, Joshi JM. Venous thromboembolism due to hyperhomocysteinaemia and tuberculosis. Natl Med J India 2017;30:139-141 |
Abstract
An 18-year-old male presented to our hospital with complaints of episodic abdominal pain, dry cough and right pleuritic chest pain. He was diagnosed as a case of right tuberculous pleural effusion on the basis of the pleural fluid Genexpert report of Mycobacterium tuberculosis detected sensitive to rifampicin and was started on antituberculous therapy. Forty-five days later, he presented with acute onset breathlessness, swelling of the right leg, streaky haemoptysis and a fresh left-sided pleural effusion. Evaluation revealed venous thromboembolism (right lower lobar segment pulmonary embolism with right leg deep vein thrombosis). Workup for malignancy was negative. However, he had vitamin B12 deficiency with increased homocysteine levels and heterozygous mutation of the MTHFR gene at A1298C. He was treated with optimal anticoagulation, vitamin B12 supplementation and antitubercular treatment. This is a rare combination of events perhaps related to the MTHFR gene mutation.
Introduction
Tuberculosis and hyperhomocysteinaemia are both pro-coagulant states. Acquired and genetic factors predispose to hypercoagu-lablitity, endothelial injury and stasis that cause thrombosis. It has also been reported as a rare systemic complication of tuberculosis due to systemic inflammatory changes causing hypercoagulability. The occurrence of an acquired and inherited factor in the same patient causing thrombosis is a rare coincidence. We describe such a patient.
The Case
An 18-year-old male presented to our outpatient department with complaints of dry cough since 2 months and recent onset right pleuritic chest pain for 15 days. Examination was normal except for signs of right pleural effusion. Biochemical investigations were within normal limits. Chest X-ray showed right subpulmonic effusion. Computed tomography (CT) of the thorax revealed right moderate pleural effusion. Aspirated pleural fluid was straw-coloured and routine biochemical evaluation suggested exudative effusion (proteins of 5.4 g/dl and total cell count 3060/cmm) with lymphocyte predominance and raised adenosine deaminase of 140 U/L. Pleural fluid Genexpert (GXP) detected low sensitivity of rifampicin to Mycobacterium tuberculosis (MTB). A diagnosis of right-sided tuberculous pleural effusion was made and the patient was started on antitubercular therapy.
One and a half months later, he presented with fresh symptoms of acute onset dyspnoea on exertion, increased dry cough with one episode of streaky haemoptysis and right leg swelling/tenderness since 4–5 days. On examination, the patient had tachycardia (pulse rate 124/minute), tachypnoea (respiratory rate 28/minute), blood pressure 120/80 mmHg, oxygen saturation 97%. He was pale, had swelling of the right leg extending to the knee, and pain and tenderness of the calf muscle. On examination of the chest, he had an impaired percussion note with shifting dullness and diminished breath sounds on the left side. The modified Wells' criteria score was 4. A provisional diagnosis of pulmonary thromboembolism (PTE) with left pleural effusion was made. Chest X-ray confirmed the left moderate pleural effusion [Figure - 1]). Evaluation for PTE revealed a raised D-dimer (1.4 mg/L) and CT pulmonary angiography showed right lower lobe segmental pulmonary embolism [Figure - 2] with a wedge-shaped area of infarction in the right posterior basal segment with left pleural effusion. The lower limb venous doppler showed right common femoral vein and popliteal vein thrombus. The two-dimensional echocardiography was normal with no evidence of right atrial/ ventricular dilatation or dysfunction. The modified pulmonary embolism severity index was 1, classified as intermediate low-risk group.
![[Figure - 1]](#fig_NatlMedJIndia_2017_30_3_139_215165_f1.jpg){kind=link}
![[Figure - 2]](#fig_NatlMedJIndia_2017_30_3_139_215165_f2.jpg){kind=link}
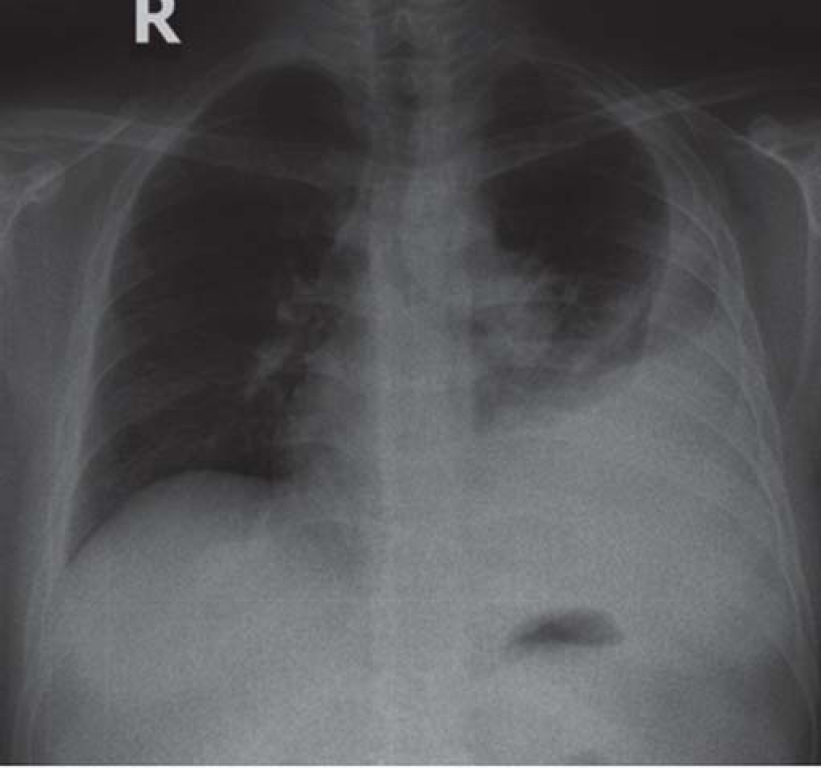 |
| Figure 1: Chest X-ray showing moderate left pleural effusion |
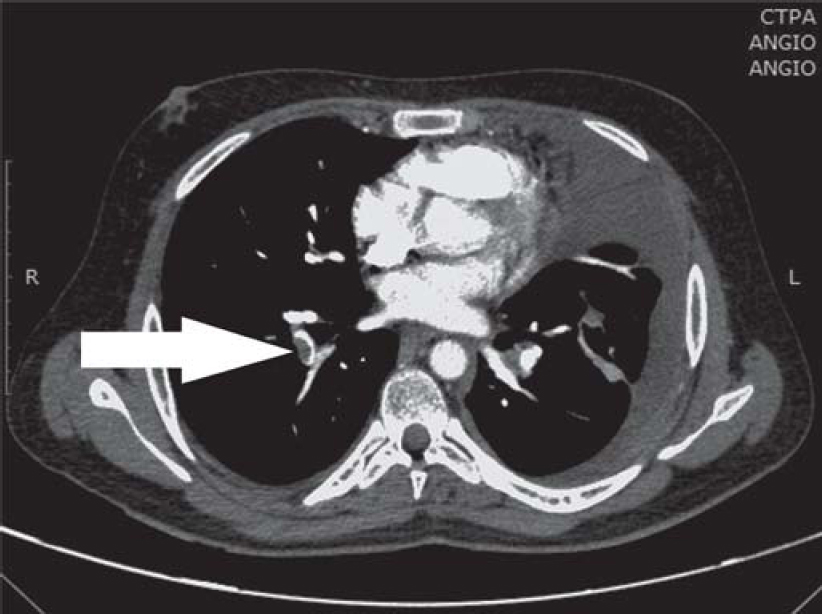 |
| Figure 2: Computed tomography pulmonary angiography (CTPA) showing right lower lobe segmental artery thrombus (arrow) |
As per the European Society of Cardiology guidelines, the patient was started on a therapeutic dose of low molecular weight heparin (enoxaparin 1.5 mg/kg/day), which was overlapped with warfarin to attain an international normalized ratio (INR) in the therapeutic range of 2–3. The pleural fluid aspirated was haemorrhagic due to PTE and was treated with pigtail drainage. Evaluation for anaemia (haemoglobin 8.4 g/dl) revealed megaloblastic anaemia with low vitamin B12 (159 pg/ml) and normal serum iron, ferritin and transferrin saturation. The serum homocysteine levels were increased (23.56 μmol/L). Further assessment with mutation analysis detected a heterozygous mutant (reduced enzyme activity) for MTHFR A1298C. A diagnosis of venous thromboembolism (VTE) due to hyperhomocysteinaemia and tuberculosis was made. He was treated with vitamin B12 supplementation. During the second hospitalization, the patient also developed recurrent right pleural effusion, which was managed with therapeutic aspiration. The swelling of the leg had initially decreased but a few days later reappeared, due to post-thrombotic syndrome, and was managed with compression stockings and limb elevation.
Discussion
VTE is a disease that includes both DVT and PTE. Hyper-coagulability, endothelial injury and stasis form the Virchow's triad, which predisposes to thrombosis. Among hospitalized patients with acute medical illness, infection, age >75 years, cancer and a history of VTE are associated with an increased risk of VTE. Long-established and well-known cardiovascular risk factors including hypertension, diabetes mellitus, smoking and high cholesterol levels have also been linked to acute PTE. Genetic risk factors for VTE include factor V Leiden, prothrombin gene mutation G20210A, protein C and S deficiency, hyperhomocysteinaemia, antithrombin deficiency and MTHFR gene C677T and A1298C mutations.[1]
Pulmonary tuberculosis causes a variety of local complications, some of which may be life-threatening. However, systemic haematological complications reported rarely with tuberculosis are disseminated intravascular coagulation (DIC) and DVT. Studies have shown subtle changes in blood rheological properties and in the haemostatic system in patients with tuberculosis. A study[2] showed erythrocyte oedema, their rapid depletion, lower resistance and higher aggregation, which is accompanied by increased haematocrit, normal erythrocyte count, increase in activated partial thromboplastin time (APTT) and thrombin time, a reduction in the values of the prothrombin indices and antithrombin III activity as possible mechanisms in tuberculosis.[3] The incidence of DIC in culture-proven tuberculosis is 3.2%, but the mortality in these patients is high (63%).[4] Robson et al.[5] reported an 8.8% incidence of DVT by venography in patients with active pulmonary tuberculosis. There have also been isolated reports of DIC due to antituberculous drugs, probably rifampicin.[6] White[7] reported that rifampicin increased 4.74-fold the risk of DVT in patients with pulmonary tuberculosis in comparison with other regimens, and DVT usually occurred within 2 weeks of starting treatment. In our patient, VTE occurred after 6 weeks of therapy. It is also postulated that the association between inflammation and haemostatic changes arising in pulmonary tuberculosis can result in a hypercoagulable state, which may predispose to DVT. Isoniazid-related B6 deficiency may be another plausible cause for hyperhomocysteinaemia, a risk factor for VTE. Cases of DVT have been reported in patients with intra-abdominal lympha-denopathy of tubercular aetiology.[8] High frequency of anti-phospholipid antibodies and deficiency of protein S in patients with tuberculosis is also mentioned in the literature.[9] In critically ill patients with tuberculosis, the degree and rate of spontaneous and stimulated platelet aggregation are decreased, which perhaps creates an additional prerequisite for progression of micro-thrombogenesis.
Hyperhomocysteinaemia, another prothrombotic condition, was present in our patient. It is a well known risk factor for coronary artery disease, myocardial infarction, venous thrombosis, cerebral infarct and PTE. The common causes of hyper-homocysteinaemia include genetic disorder, dietary deficiency of folic acid, vitamin B12 or vitamin B6, chronic renal insufficiency, lifestyle factors (chronic alcohol intake, smoking or high coffee intake), end-stage diabetes, systemic lupus erythematosus, hyperproliferative disorders and medications (methotrexate, sulphonamides or antacid). Many hypotheses have been proposed to explain how hyperhomocysteinaemia may lead to venous thrombosis and atherosclerosis. One such hypothesis is that homocysteine has a toxic effect on the vascular endothelium and the clotting cascade.[10] Alternatively, hyperhomocysteinaemia may reflect abnormal methionine metabolism that affects the methylation of DNA and cell membranes. Our patient had associated anaemia due to vitamin B12 deficiency. Elevated homocysteine levels may result from low levels of folic acid, vitamin B6 or vitamin B12.[11] Moreover, several genetic alterations in enzymes involved in homocysteine metabolism have been described. Currently, over 40-point mutations of this gene have been identified. Of these, mutations on the points at C677T and A1298C seem to have the most clinical significance. Our patient had heterozygous mutant detected for MTHFR A1298C, which implies reduced enzyme activity associated with folate metabolism. The A1298C polymorphism is characterized by a point mutation at position 1298 in exon 7 of the MTHFR gene that leads to replacement of glutamine by alanine in the enzyme. Individuals homozygous for the A1298C allele do not show higher serum homocysteine levels than controls. However, MTHFR activity is reduced and homocysteine levels are increased in those heterozygous for both the A1298C and C677T polymorphisms and in individuals homozygous for the C677T polymorphism alone. It remains unclear whether hyperhomocysteinaemia due to different causes has the same risk of thrombosis.[12] The MTHFR gene mutation in our patient may be a coincidental finding and thromboembolism may or may not have been a result of the mutation. Nevertheless, it is well known that vitamin supplementation lowers homocysteine concentrations in almost all patients with hyperhomocysteinaemia, regardless of the underlying cause. Naurath et al.[13] reported that high-dose multivitamins, including folate, vitamin B6 and vitamin B12, resulted in a 49.5% reduction in the mean homocysteine level. den Heijer et al. reported that combined supplementation with folate, cobalamin and pyridoxine reduced homocysteine levels by 30% within 8 weeks in patients with recurrent venous thrombosis.[14]
Tuberculosis with hyperhomocysteinaemia is postulated as the cause for VTE in our patient. We did not come across this rare concurrence in the literature.
| 1. | Osinbowale O, Ali L, Chi YW. Venous thromboembolism: A clinical review. Postgrad Med 2010; 122:54–65. [Google Scholar] |
| 2. | Kaminskaia GO, Serebrianaia BA, Martynova EV, Mishin VI. Intravascular coagulation as a typical concomitant of acute pulmonary tuberculosis. Probi Tuberk 1997; 3:42–6. [Google Scholar] |
| 3. | Wang JY, Hsueh PR, Lee LN, Liaw YS, Shau WY, Yang PC, et al. Mycobacterium tuberculosis inducing disseminated intravascular coagulation. Thromb Haemost 2005;93:729–34. [Google Scholar] |
| 4. | Kaul S, Zadeh AA, Shah PK. Homocysteine hypothesis for atherothrombotic cardiovascular disease: Not validated. J Am Coll Cardiol 2006;48:914–23. [Google Scholar] |
| 5. | Robson SC, White NW, Aronson I, Woolgar R, Goodman H, Jacobs P. Acute-phase response and the hypercoagulable state in pulmonary tuberculosis. Br J Haematol 1996;93:943–9. [Google Scholar] |
| 6. | IP M, Cheng KP, Cheung WC. Disseminated intravascular coagulopathy associated with rifampicin. Tubercle 1991;72:291–3. [Google Scholar] |
| 7. | White NW. Venous thrombosis and rifampicin. Lancet 1989;2:434–5. [Google Scholar] |
| 8. | Gogna A, Pradhan GR, Sinha RS, Gupta B. Tuberculosis presenting as deep vein thrombosis. Postgrad Med J 1999;75:104–5. [Google Scholar] |
| 9. | Suarez Ortega S, Artiles Vizcaino J, Balda Aguirre I, Melado Sanchez P, Arkuch Saade ME, Ayala Galan E, et al. Tuberculosis as risk factor for venous thrombosis. An Med Interna 1993;10:398^00. [Google Scholar] |
| 10. | Blom HJ, VanDer Molen EF. Pathobiochemical implications of hyperhomo-cysteinemia. Fibrinolysis 1994;8:86–7. [Google Scholar] |
| 11. | Engbersen AMT, Franken DG, Boers GHJ, Stevens EMB, Trijbels FJM, Blom HJ. Thermolabile 5, 10-methylenetetrahydrofolate reductase as a cause of mild hyperhomocysteinemia. Am J Hum Genet 1995;56:142–50. [Google Scholar] |
| 12. | den Heijer MD, Brouwer IA, Bos GJ, Blom HJ, van der Put NJ, Spaans AP, et al. Vitamin supplementation reduces blood homocysteine levels: A controlled trial in patients with venous thrombosis and healthy volunteers. Arterioscler Thromb Vasc Biol 1998;18:356–61. [Google Scholar] |
| 13. | Naurath HJ, Joosten E, Riezler R, Stabler SP, Allen RH, Lindenbaum J. Effects of vitamin B12, folate, and vitamin B6 supplements in elderly people with normal serum vitamin concentrations. Lancet 1995;346:85–9. [Google Scholar] |
| 14. | den Heijer M, Willems HP, Blom HJ, Gerrits WBJ, Cattaneo M, Eichinger S, et al. Homocysteine lowering by B vitamins and the secondary prevention of deep vein thrombosis and pulmonary embolism: A randomized, placebo-controlled, doubleblind trial. Blood 2007;109:139^4. [Google Scholar] |
Fulltext Views
1,402
PDF downloads
398



