Translate this page into:
A comparative study of the informed consent process with or without audiovisual recording
Corresponding Author:
U M Thatte
Department of Clinical Pharmacology, First Floor, New Multistorey Building, Seth G.S. Medical College and K.E.M. Hospital, Parel, Mumbai 400012, Maharashtra
India
urmilathatte@gmail.com
| How to cite this article: Figer B H, Chaturvedi M, Thaker S J, Gogtay N J, Thatte U M. A comparative study of the informed consent process with or without audiovisual recording. Natl Med J India 2017;30:262-265 |
Abstract
Background. The Central Standard Drugs Control Organization (CDSCO) issued an administrative order in November 201 3 mandating audiovisual (AV) recording of the informed consent process for all regulatory studies. At this point, a phase 2/3 trial ongoing at our centre had recruited 45 participants using the written, informed consent process. Another 40 participants were recruited after the order and underwent AV recording of the consent process. We assessed the difference in participants’ understanding between the two consenting processes as the trial fortuitously had both forms of consent.Methods. A 16-item questionnaire with six domains (purpose, study procedures, risks, benefits, payment for participation, and rights and confidentiality) was designed and validated. It was administered to the participants after approval of the institutional ethics committee and written informed consent. Answers given were matched with a template of model answers. The responses were scored as fully correct (3), partially correct (2), ‘don't remember’ (1 ), and incorrect (0) with a total possible score of 48. Between-group analysis was done for total scores and domain-specific scores. Domain-wise analysis was done for the proportion of all categories of responses. The impact of potential confounders on participants’ understanding was also factored in.
Results. A total of 38 respondents—21 in the AV consent group and 1 7 in the written consent group—agreed to participate. The total mean (SD) score of the AV consent group was significantly higher (40.3 [5.9]) compared to that of the written consent group (34.8 [7.94]; p = 0.01). Between the groups the score was significant in the domains of rights and confidentiality (p = 0.01). The proportion of participants who gave fully correct answers was statistically significant in the domain of purpose (p = 0.04). The time elapsed between the original consent and this study showed a weak inverse correlation (ρ = -0.3, p = 0.01).
Conclusion. AV recording of the informed consent process in a clinical trial appears to improve the understanding of participants relative to the written informed consent alone.
Introduction
The informed consent process has its origins in the Nuremberg Code and forms the cornerstone of clinical research.[1] Obtaining written, informed consent from participants before initiating research is a universally accepted norm. The Declaration of Helsinki (2013) states that investigators should enrol participants only after they have ascertained that ' they have understood’' what the study entails.[2] In practice, investigators are rarely likely to determine with certainty the ‘understanding’ by participants and the voluntariness of their decision-making.[3]
The Central Drugs Standard Control Organization (CDSCO), the regulatory agency responsible for clinical trials, issued an administrative order on 19 November 2013 mandating the audiovisual (AV) recording of the informed consent process. The notification stipulated that ‘in addition to the requirement of obtaining written, informed consent, audiovisual recording of the informed, consent process of each trial subject, including the procedure of providing information to the subject and his/her understanding on such consent is required to be done while adhering to the principles of confidentiality’.[4]
At the point of this notification, a clinical trial (CTRI/2012/05/ 002709) investigating an antirabies monoclonal antibody was ongoing at our centre. Till then, 45 patients had been recruited after giving written informed consent. Subsequent to the notification, which mandated AV recording of the consent process, 40 more patients were enrolled. Since a single trial fortuitously had participants who had undergone both consenting processes, we did this study to assess whether there was a difference in the understanding of the trial participants between the two consenting processes.
Methods
Ethics
The approval of the institutional ethics committee was taken and written, informed consent obtained from all the participants.
Setting
The study was conducted in the Department of Clinical Pharmacology of Seth G.S. Medical College and K.E.M. Hospital, Mumbai.
Selection of participants
All those who consented to take part in the ongoing phase 2/3 rabies monoclonal antibody trial were eligible to participate.
Intervention
Development and validation of questionnaire. Participants’ understanding of the trial was assessed using a 16-item questionnaire (available at www.nmji.in) consisting of the following six domains (based on the draft guidelines[5] for obtaining AV consent issued by CDSCO): (i) purpose of the trial; (ii) procedures to be followed; (iii) risks; (iv) benefits; (v) compensation; and (vi) rights and confidentiality.
Thirteen experts in ethical and regulatory aspects of clinical trials, each of whom had a minimum of 5 years’ experience in this area, evaluated the content validity for the questionnaire independently and rated every question as either ‘essential’, ‘useful’ or ‘not needed’. Those questions rated as ‘essential’ or ‘useful’ were retained and modified. Lawshe's content validity ratio (CVR) was calculated. All questions that had a CVR of >0.54 were retained in the final questionnaire.[6]
The test-retest reliability was assessed by administering the questionnaire to 10 normal, healthy participants and calculating the inter-class and intra-class correlation coefficients. Cronbach’ s alpha was calculated as a measure of internal consistency.
The translations in regional languages, i.e. Hindi and Marathi, were authenticated by a language expert and back translation certificates obtained. The translations were further validated and assessed for reliability.
A template of correct answers was developed and scores were assigned to the participants’ response as follows: fully correct: 3; partially correct: 2; can't say/don't remember: 1 ; and incorrect: 0. The maximum possible score for the 16-item questionnaire was 48.
Administration of the questionnaire. The questionnaire was administered by any one of the three team members (BF, MC and ST) of the trial after obtaining written, informed consent from each participant. All participants were given adequate time to complete the questionnaire and if they had any doubts these were clarified by one of the team members.
Primary outcome of interest
Calculation of scores. Answers given by the participants were matched with a template of correct answers and scores. All disputes arising while assigning the scores were resolved through discussion with senior authors NJG and UMT. The total and domain-specific scores were calculated for each participant.
The proportion of participants who gave fully correct (scored 3), partially correct (scored 2) or fully incorrect response (scored 0) and those who replied ‘cannot say/do not remember’ (scored 1) for each question was recorded.
To identify the proportion of participants who gave each of these subtype responses ‘domain-wise’, we devised a scoring system to account for the fact that each domain had more than one question and each participant could give any of the four responses or a combination of responses. The maximum total score possible for each domain (if fully correct answers were given for all the que stions in that domain) was calculated—purpose : 6 (2 que stions) ; procedures: 12 (4 questions); risks: 9 (3 questions); payment for participation: 3 (1 question); benefits: 6 (2 questions); and rights and confidentiality: 12 (4 questions). The per cent (%) score obtained for each domain was then calculated for each participant. Participants scoring 75% or more for a particular domain were categorized as ‘having good understanding’, those who scored between 60% and 74% as having reasonable understanding and those scoring less than 60% as ‘inadequate understanding’ for that domain. For example, in the domain of purpose, the maximum possible score was 6. Thus, a participant scoring either 5 or 6 (75% or more) was categorized as having given a ' fully correct’ response, 4 as ‘partially correct’, and 3 or lower as ‘incorrect’. This scoring was done post hoc for ease of interpretation.
Secondary outcomes of interest
Time taken to administer consent. According to the departmental standard operating procedures and as part of the data collected in this regulatory trial, the duration of the informed consent process in the two groups was noted.
Addressing the confounders. The following potential confounders were assessed for their influence on the total score: time elapsed (in number of days) from the date of the informed consent given for the original interventional trial until the administration of the questionnaire, literacy, socioeconomic class (as assessed by the Kuppuswamy scale[7]), and age of the participants. The duration of the consent process (obtained from source notes) for each participant was also recorded.
Statistical analysis
Demographics were assessed using descriptive statistics. The study sample comprised all eligible participants (45 in the written informed consent group and 40 in the AV recording group). The scores were assessed for normality using the Shapiro-Wilk test. The difference in total scores, the difference in time elapsed since the original consent and duration of consent procedure between the groups were assessed using the Mann–Whitney U test. The Chi-square test for trend was used to compare the proportion of participants who gave fully correct, partially correct or fully incorrect responses domain-wise. The ‘cannot say/do not remember’ were clubbed with the ‘incorrect responses’ for the purpose of analysis. The effect of potential confounders was assessed in a univariate analysis using Spearman's rho (ρ) and the coefficient of determination was calculated. The analysis was performed using GraphPad InStat version 3.0 software. Significance was considered at p<0.05.
Results
Demographics
Of the 85 eligible participants, only 38 from each group could be contacted. Of them, 17 of 38 in the written informed consent group and 21of 38 in the AV consent group agreed to participate, giving a consent decline rate of 50%. The primary reasons cited by those who declined consent to participate in this study were primarily three: lack of time, not interested, and left the city.
No between-group difference was seen with respect to age, gender, literacy, and socioeconomic class [Table - 1]. A statistically significant difference however was seen between the two groups with respect to the duration of the original consent process as also the time elapsed between the original trial and the present study.
![[Table - 1]](#tbl_NatlMedJIndia_2017_30_5_262_234392_t1.jpg){kind=link}

Questionnaire validation
The overall CVR was 0.77 and domain-specific CVRs ranged from 0.65 to 1, with the lowest CVR score for the domain of rights and confidentiality and highest for the domain of compensation. Overall inter-class and intra-class correlation coefficients were 0.91 and 0.80, respectively and the Cronbach's alpha was 0.7.
Total and domain-specific scores
The total score in the AV consent group was significantly higher than that in the written consent group (p=0.01 ; [Table - 2]. The AV consent group also had higher domain scores (than the written consent group) in all domains except that of benefits. However, the difference reached statistical significance (p=0.01) only in the domain of rights and confidentiality [Table - 2].
![[Table - 2]](#tbl_NatlMedJIndia_2017_30_5_262_234392_t2.jpg){kind=link}

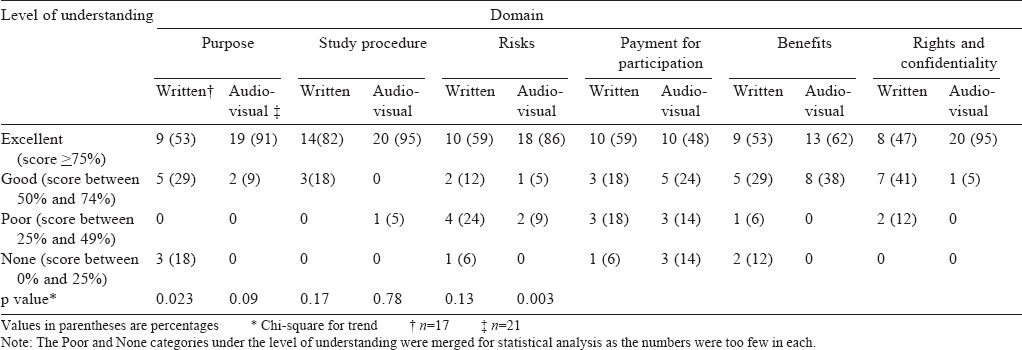
Analysis of domain-wise understanding between the groups A statistically significant difference in understanding was found between the two groups in two domains: that of purpose (p=0.023) and rights and confidentiality (p=0.003) with the AV consent group showing a better understanding. [Table - 3] gives the details of domain-wise responses of the participants.
![[Table - 3]](#tbl_NatlMedJIndia_2017_30_5_262_234392_t3.jpg){kind=link}
Impact of confounders
The mean (SD) time elapsed (in number of days) from the date of the informed consent given for the original interventional trial until the administration of the questionnaire was 828 ( 186.8) days for the written informed consent group versus 449 (137.1) days for the AV consent group (p=0. 02). A weak association was found between this time and the total scores obtained r=-0.4 (p=0.007). None of the other variables showed a significant association (literacy, r=-0.202, p=0. 22 ; socioeconomic class, r=-0. 12, p=0. 5 ; or age r=-0.189, p=0.255). Although the mean (SD) time taken to administer consent was significantly (p<0.05) greater in the AV consent group 68 (30.77) compared to the written consent group 50 (19.55), there was no association between the time for consent and the total score (r=0.24, p=0.15).
Coefficient of determination
The rho (ρ) of -0.4 yielded a coefficient of determination of 0.16 indicating that only 16% of the variance in the total score could be explained by recall bias.
Discussion
The principle of respect for individuals’ rights requires that those who participate in research be provided with sufficient information that they understand, to make autonomous and informed decisions about whether or not to consent to participate. However, evidence overwhelmingly suggests that participants’ comprehension about the research they participate in is often poor.[8],[9] We found that the use of AV recording resulted in overall better comprehension as also higher scores in five of the six domains tested. A larger proportion of participants in the AV consent group also gave fully correct answers to the questions.
The metrics used by studies in the literature to assess comprehension of consent have largely been based on questionnaires. Within the questionnaire, testing options include multiple-choice questions and true or false statements.[9] We too used a questionnaire (Appendix 1 ; available at www.nmji.in) that was validated and translated into two local languages. It had a mix of open-ended and multiple-choice questions and covered the important elements which are recommended by ethics guidelines to be part of consent.
We found that the AV recording process enhanced understanding about the study significantly as seen by better total scores in that group, a greater proportion of participants scoring >80% as well as a higher proportion of fully correct answers and a lower proportion of partially correct and incorrect answers in the AV consent group as compared to the written consent group.
A systematic review evaluated the papers published between 1966 and March 2004[10] on the interventions used to improve research participants’ understanding of the consent process. The authors classified interventions into five broad categories: multimedia, enhanced consent forms, extended discussion, test/ feedback, and miscellaneous. All categories of interventions showed mixed results indicating that none of the interventions by themselves improved research participants’ understanding completely. However, another similar systematic review, a decade later which also included a meta-analysis,[11] had slightly different results. It showed that among all interventions studied, enhanced consent forms and extended discussions were most effective in improving participants’ understanding. Our study could perhaps be categorized as an ' extended discussion’ intervention as evinced by the fact that those who underwent AV recording of the consent process spent on an average 20 minutes longer than those who did not.
We found that participants’ understanding varied across all domains with significantly better understanding seen in the domains of purpose and rights and confidentiality. The domains in the questionnaire were created on the basis of the guidelines[5] for obtaining AV consent issued by regulatory authorities. These guidelines have an entire section devoted to the issue of privacy and confidentiality. This section explains that the person administering the consent should pay special emphasis on this issue so that the patient gains a better understanding. In the domain of benefits, there was generally a poor understanding or recall (a score of about 50% in both groups). Our study was not designed to understand the ‘why’ of ‘benefits’. However, while assessing benefits (or risks) it is important to consider the patient's perspective.[12] Since the consent process emphasized that as this was a research trial, and the participant may not ‘get benefit’, this could have led to the perception that there was no benefit to be expected; thus a low score in this domain.
Several factors have been identified that can affect comprehension of informed consent. These include: the level of education, the extent of information an individual can process,[13] length and complexity of the informed consent form, hope for clinical benefit (therapeutic mis-estimation), therapeutic misconception, and reduction in ability to remember study information over time.[14],[15] We assessed six factors for their confounding potential—literacy, age, gender, socioeconomic strata, time to administer consent, and time elapsed from the consent given for the original trial until the point of administering the questionnaire. Barring the last factor, none impacted the difference observed between the two groups. Those who underwent AV consent as expected had a smaller and statistically significant time gap between the administration of their original consent to the trial and the present study. However, the coefficient of determination of 16% derived from the rho indicated that the time elapsed accounts for only 16% of the variability in the total scores.
Limitations
The fact that a single trial fortuitously had both consenting processes is both a strength and a weakness. It would be extremely difficult if not impossible for researchers in India to find studies that have only the AV consent process and match them for all variables with those that have only the written informed consent process. This is also a limitation as the AV consent happened subsequent to the gazette notification and after the written informed consent process.
Conclusion
The process of capturing the informed consent process on tape and camera appears to result in a better understanding of most elements of the consent process.[16]
| 1. | The Nuremberg Code. Available at www.hhs.gov/ohrp/archive/nurcode.html (accessed on 7 Apr 2016). [Google Scholar] |
| 2. | WMA Declaration of Helsinki—Ethical Principles for Medical Research Involving Human Subjects. Available at www.wma.net/en/30publications/10policies/b3/ (accessed on 9 Apr 2016). [Google Scholar] |
| 3. | Wendler D. How to enroll participants in research ethically. JAMA 2011;305: 1587-8. [Google Scholar] |
| 4. | Order. Directorate General of Health Services, Ministry of Health and Family Welfare, Office of Drug Controller General India. 19 Nov 2013. Available at www. cdsco. nic. in/writereaddata/Office%20Order%20dated%2019.11.2013.pdf (accessed on 2 Apr 2016). [Google Scholar] |
| 5. | Draft guidelines on audio-visual recording of informed consent process in clinical trial. Central Drugs Standard Control Organization. Directorate General of Health Services. Ministry of Health and Family Welfare, Government of India. 9 Jan 2014. Available at www. cdsco. nic. in/writereaddata/Office%20Order%20dated%2019. 11.2013.pdf (accessed on 17 Aug 2016). [Google Scholar] |
| 6. | Ayre C, Scally AJ. Critical values for Lawshe’ s content validity ratio : Revisiting the original methods of calculation. Meas Eval Couns Dev 2014;47:79-86. [Google Scholar] |
| 7. | Sharma R. Kuppuswamy's socioeconomic status scale—revision for 2011 and formula for real-time updating. Indian J Pediatr 2012;79:961-2. [Google Scholar] |
| 8. | Afolabi MO, Okebe JU, McGrath N, Larson HJ, Bojang K, Chandramohan D. Informed consent comprehension in African research settings. Trop Med Int Health 2014;19:625-42. [Google Scholar] |
| 9. | Dunn LB, Jeste DV. Enhancing informed consent for research and treatment. Neuropsychopharmacology 2001;24:595-607. [Google Scholar] |
| 10. | Kass NE, Taylor HA, Ali J, Hallez K, Chaisson L. A pilot study of simple interventions to improve informed consent in clinical research: Feasibility, approach, and results. Clin Trials 2015;12:54-66. [Google Scholar] |
| 11. | Flory J, Emanuel E. Interventions to improve research participants’ understanding in informed consent for research: A systematic review. JAMA 2004;292:1593-601. [Google Scholar] |
| 12. | Nishimura A, Carey J, Erwin PJ, Tilburt JC, Murad MH, McCormick JB. Improving understanding in the research informed consent process: A systematic review of 54 interventions tested in randomized control trials. BMC MedEthics 2013;14:28. doi: 10.1186/1472-6939-14-28 (accessed on 7 Apr 2016). [Google Scholar] |
| 13. | Zvonareva O, Kutishenko N, Kulikov E, Martsevich S. Risks and benefits of trial participation: A qualitative study of participants’ perspectives in Russia. Clin Trials 2015;12:646-53. [Google Scholar] |
| 14. | Davis TC, Holcombe RF, Berkel HJ, Pramanik S, Divers SG. Informed consent for clinical trials: A comparative study of standard versus simplified forms. J Natl Cancer Inst 1998;90:668-74. [Google Scholar] |
| 15. | Chaisson LH, Kass NE, Chengeta B, Mathebula U, Samandari T. Repeated assessments of informed consent comprehension among HIV-infected participants of a three-year clinical trial in Botswana. PLoS One 2011;6:e22696. doi:10.1371/journal.pone. 0022696. [Google Scholar] |
| 16. | Olver IN, Buchanan L, Laidlaw C, Poulton G. The adequacy of consent forms for informing patients entering oncological clinical trials. Ann Oncol 1995;6:867-70. [Google Scholar] |
Fulltext Views
2,898
PDF downloads
1,134



