Translate this page into:
Drooping shoulders: A rare manifestation of an uncommon disease
2 Department of Radiodiagnosis, All India Institute of Medical Sciences, New Delhi 110029, India
Corresponding Author:
Rishav Bansal
Department of Geriatric Medicine, All India Institute of Medical Sciences, New Delhi 110029
India
rishavbansal.aiims@gmail.com
| How to cite this article: Bansal R, Singh S, Singhal S, Dewangan G, Das CJ, Dey AB. Drooping shoulders: A rare manifestation of an uncommon disease. Natl Med J India 2020;33:276-277 |
Abstract
Primary systemic amyloidosis is an uncommon disease which presents with involvement of the kidney, heart, peripheral nervous system and liver. The involvement of skeletal muscles called amyloid myopathy is uncommon. We report a 74-year-old male who presented with progressively drooping shoulders followed by other muscular involvement without other organ involvement as a manifestation of amyloidosis. The patient was managed with melphalan, prednisolone and thalidomide with clinical improvement.Introduction
Primary systemic amyloidosis (AL) is a systemic disease with a reported incidence of <8–13 cases per million per year.[1] The usual manifestations are nephrotic syndrome, peripheral neuropathy, hepatomegaly and skin involvement.[2] The involvement of muscle tissue in the form of cardiomyopathy or macroglossia is not uncommon. However, amyloid myopathy (AM) presenting as progressive proximal weakness and increased creatinine phosphokinase (CPK) level is rarely reported.[3],[4],[5] AM presenting with axial musculature has not been reported. We report a patient of primary systemic AL presenting with paraspinal muscle involvement leading to stooping posture without any other organ involvement.
The Case
A 74-year-old male with obstructive sleep apnoea for 2 years presented with gradually progressive forward drooping of the cervicodorsal spine on walking for the preceding 18 months. His forward drooping increased progressively with time from after about 100 steps to the first step, which made walking almost impossible. Over time, he became home- and chair-bound and then bed bound most of the time.
After 6 months of axial muscle involvement, he developed additional symptoms of progressive difficulty in climbing stairs, combing hair, drooping of eyelids, slurring of speech, difficulty in protruding his tongue, difficulty in chewing food and increased time to complete his meal. He also noticed swelling of the legs and erythematous skin lesions on both arms with peeling of the skin. He had undocumented weight loss during this period. His symptoms had no exacerbating or relieving factors. There was no diurnal variation in any of his symptoms, and the intensity of symptoms was not related to exertion or rest. He had no other comorbid condition or age-related health issue.
He was a thin built person with pallor, patches of erythematous skin lesions on his arms, bilateral ptosis, kyphosis and slurred speech. His tongue was stiff and had restricted mobility. He had bilateral drooping of eyelids, although all his cranial nerves were normal. He had weakness of proximal muscles of both upper and lower limbs (power Grade 4/5) and truncal muscles; with no atrophy, fasciculation or hypotonia. Deep and superficial reflexes, all modalities of sensation and autonomic functions were intact. All other organ systems were normal.
He had normocytic normochromic anaemia; mild hyper-calcaemia, low albumin, reversed albumin and globulin ratio; and mildly raised CPK level. He had normal concentrations of vitamin B12, folate, vitamin D and parathyroid hormone. The systemic evaluation showed microalbuminuria, negative urine active sediment and normal renal ultrasound; normal electrocardiography and normal echocardiography with elevated NT-pro BNP; negative autoimmune markers (antinuclear antibody) and negative viral markers (HIV, hepatitis B surface antigen and hepatitis C virus antibody); bilateral mild effusion on chest X-ray and axonal sensorimotor neuropathy involving all limbs on nerve conduction study with normal electromyography and negative neostigmine test. Ultrasound neck done to look for a local cause of immobile tongue was normal. X-ray of the whole spine suggested degenerative changes without any fracture with dual-energy X-ray absorptiometry scan revealing osteoporosis. CT scan revealed evidence of infiltration of fat into the pericardium and diffuse atrophy of the neck muscles particularly the posterior cervical muscles showing diffuse fatty infiltration. Magnetic resonance imaging of the spine revealed heterogeneous signal intensity in both T1 and T2 images in the paraspinal muscles of the neck [Figure - 1]..
![[Figure - 1]](#fig_NatlMedJIndia_2020_33_5_276_317479_f1.jpg){kind=link}
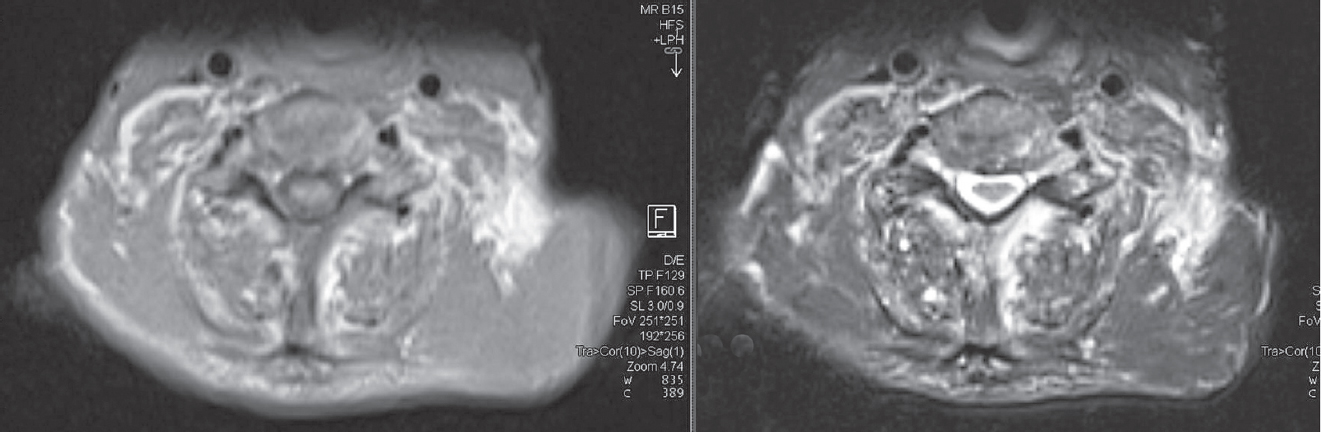 |
| Figure 1: Magnetic resonance imaging of the spine reveals heterogeneous signal intensity in paraspinal muscles |
In view of reversal of the albumin to globulin ratio, mild hypercalcaemia, mild anaemia and multi-organ involvement, work-up for multiple myeloma and AL was done. Serum and urine electrophoresis revealed dense M band in α-gamma mobility area, serum immunofixation revealed M spike as IgG, lambda and its isomer lambda, a2-microglobulin level of 5.76 mg/L, serum-free light chain assay with λ chains level of >1650 mg/L (normal range 5.7–26.3 mg/L) and κ chains level of 33.82 mg/L (normal range 3.3–19.4 mg/L), skeletal survey revealed no lytic lesion and bone marrow biopsy confirmed the presence of plasma cells present in significant amount [Figure - 2]. Abdominal fat pad biopsy also confirmed AL [Figure - 3]. A final diagnosis of primary systemic AL type due to multiple myeloma was made. Further, biopsy of sural nerve and muscle was declined by the patient as diagnosis had already been made. The patient was started on melphalan, prednisolone and thalidomide (MPT) and after 1 month, there was an improvement in tongue mobility with improved speech and improvement in muscle power. He was surviving when last followed up 6 months after treatment.
![[Figure - 2]](#fig_NatlMedJIndia_2020_33_5_276_317479_f2.jpg){kind=link}
![[Figure - 3]](#fig_NatlMedJIndia_2020_33_5_276_317479_f3.jpg){kind=link}
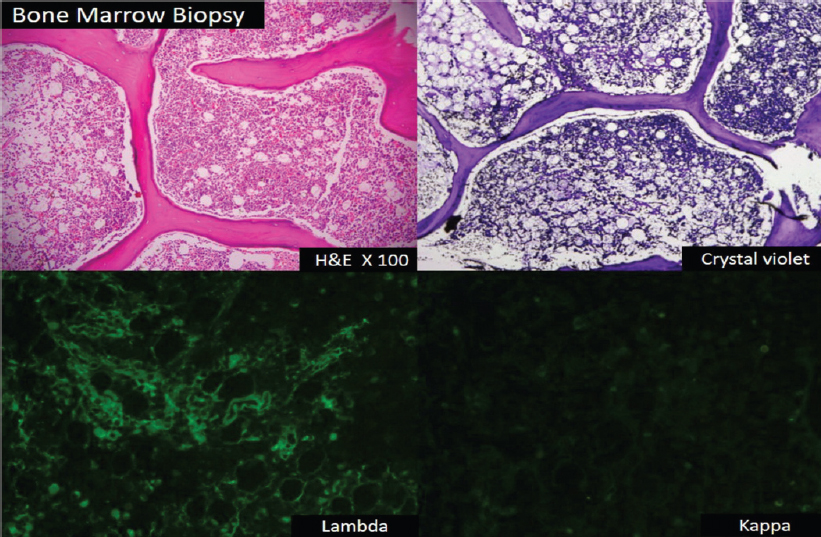 |
| Figure 2: Multiple myeloma and amyloidosis in bone marrow biopsy |
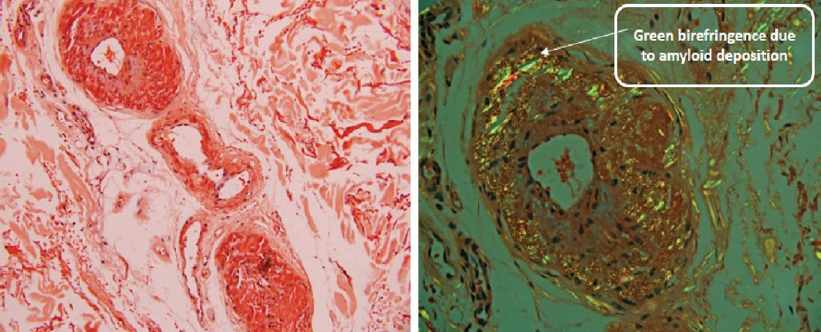 |
| Figure 3: Amyloidosis in abdominal fat pad biopsy (Congo red stain seen under polarized microscopy) |
Discussion
Although AM is a well-described disorder, it is often overlooked for two reasons: first, it is rarely observed clinically, and second, a broad span of differential diagnoses is needed. Furthermore, it may be worth noting that the predominant manifestation initially may be of proximal muscle weakness and other manifestations due to amyloid deposition at other sites may develop only subsequently. In this case, the patient presented with myopathy involving axial muscles first with progression to involve proximal limb muscles and other organ systems later.
AM usually occurs in patients with primary AL type, which was the case in our patient. AL accounts for the vast majority of reported cases of AM.[6] Physical examination classically reveals macroglossia or pseudohypertrophy of affected muscles. However, three of the largest case series to date have noted that <25% of patients present with pseudohypertrophy; the remaining patients have muscle atrophy or normal muscle bulk.[3] Our patient did not have macroglossia or pseudohypertrophy.
In a retrospective analysis of patients over 27 years at Mayo Clinic, only 12 patients of amyloid deposit in the muscle were seen. Their long-term prognosis was poor.[2] The median survival of primary AL is 14.7 months from diagnosis and even worse when associated with multiple myeloma with median survival of 4–5 months from diagnosis.[7] Response to cytotoxic agents is also poor. Death usually occurs due to cardiac arrhythmias or progressive cardiac failure. There have been few documented remissions of AL. Occasional patients may improve with treatment of the underlying disorder. Regression of histological evidence of amyloid and clinical remission in primary AL is rare.[8]
Conclusion
We report this atypical presentation (drooping of shoulders due to early axial muscle involvement) of an uncommon disease (AM) with good response to the MPT regimen.
Conflicts of interest. None declared
| 1. | Nienhuis HL, Bijzet J, Hazenberg BP. The prevalence and management of systemic amyloidosis in western countries. Kidney Dis (Basel) 2016;2:10–19. [Google Scholar] |
| 2. | Gertz MA, Kyle RA. Myopathy in primary systemic amyloidosis.J Neurol Neurosurg Psychiatry 1996;60:655–60. [Google Scholar] |
| 3. | Prayson RA. Amyloid myopathy: Clinicopathologic study of 16 cases. Hum Pathol 1998;29:463–8. [Google Scholar] |
| 4. | Chapin JE, Kornfeld M, Harris A. Amyloid myopathy: Characteristic features of a still underdiagnosed disease. Muscle Nerve 2005;31:266–72. [Google Scholar] |
| 5. | Tuomaala H, Kärppä M, Tuominen H, Remes AM. Amyloid myopathy: A diagnostic challenge. Neurol Int 2009; 1:e7. [Google Scholar] |
| 6. | M’bappé P, Grateau G. Osteo-articular manifestations of amyloidosis. Best Pract Res Clin Rheumatol 2012;26:459–75. [Google Scholar] |
| 7. | Glenner GG. Amyloid deposits and amyloidosis: The beta-fibrilloses (second of two parts). N Engl J Med 1980;302:1333–43. [Google Scholar] |
| 8. | Sheehan-Dare RA, Simmons AV. Amyloid myopathy and myeloma: Response to treatment. Postgrad Med J 1987;63:141–2. [Google Scholar] |
Fulltext Views
1,639
PDF downloads
344



