Translate this page into:
Management of advanced melanoma in the current era: A medical oncology perspective for the Indian scenario
Corresponding Author:
Sameer Bakhshi
Department of Medical Oncology, Dr B.R. Ambedkar Institute Rotary Cancer Hospital, All India Institute of Medical Sciences, Ansari Nagar, New Delhi 110029
India
sambakh@hotmail.com
| How to cite this article: Mittal A, Pushpam D, Bakhshi S. Management of advanced melanoma in the current era: A medical oncology perspective for the Indian scenario. Natl Med J India 2020;33:89-98 |
Abstract
Malignant melanoma is an aggressive malignancy with high recurrence rates after curative surgery and in advanced stages is characterized by resistance to conventional chemotherapy. With better understanding of the genomic landscape and mutational signature of these tumours over the past decade, there has been a paradigm shift in management of melanoma using immunotherapy (anti-PD-1 and anti-CTLA-4 antibodies) and targeted drugs against BRAF and MEK. These drugs have shown survival benefits in both adjuvant and metastatic setting with patients being eligible for immunotherapy irrespective of any biomarker. However, these drugs have varying toxicity profiles and there are no studies comparing these two classes of drugs in either the adjuvant or metastatic setting leaving the question of sequencing open to clinical judgement. Moreover, availability and cost are issues that need to be considered before use of these drugs in the Indian setting.Introduction
Melanoma is an uncommon malignancy with wider racial, ethnic and geographical variations in incidence than reported with any other cancer. Since we last reviewed the subject in 2010,[1] the treatment of melanoma has undergone a paradigm shift with the introduction of immunotherapy and targeted therapy. Management of metastatic melanoma is one of the biggest success stories of oncology; 5-year survival has jumped from <10% before 2011 with chemotherapy[2] to >40% with combination immunotherapy in 2019 with median overall survival (OS) of >2 years. Most of the data available are for cutaneous melanoma; practice in mucosal melanoma is based on extrapolation of that data. We summarize the evidence and rationale for various therapeutic approaches in advanced melanoma focusing on key phase 3 trials.
Epidemiology and Risk Factors
Age-standardized incidence rates (age-adjusted rate [AAR]) of melanoma have increased steadily over the past two decades with the highest incidence in Australia (33/100 000/year).[3],[4],[5],[6] India has one of the lowest AARs in the world (0.2/100 000/ year). The reason for this large difference in incidence in India compared with the West is understandable because of the more wheatish/brown/black skin complexion in India and underreporting as most localized disease presents to dermatologists/surgeons. Mucosal and acral melanoma have been reported more commonly in India compared to the West, though no concrete data on epidemiology of melanoma in India exist.[7],[8] At present, melanoma is not recorded in hospital- or population- based cancer registries in India. The major risk factors for melanoma include white race and exposure to ultraviolet radiation, predominantly ultraviolet B. Intense intermittent exposure to sunlight with tendency to develop sunburns has been implicated in the pathogenesis of cutaneous melanoma.[9],[10] Familial clustering is seen in 10%–15% of patients with melanoma. The most common gene associated with hereditary melanoma is CDKN2A/p16, mutation of which also leads to predisposition for pancreatic cancer; less common genes include CDK4, TERT, BAP1 and POT1.[11] Other risk factors include a personal history of melanoma, multiple naevi and atypical naevi.
Clinical Presentation and Diagnosis
Subtypes
Six clinical subtypes of melanoma have been described and each has variable presentation, ethnic differences and prognosis [Table - 1].[12]
![[Table - 1]](#tbl_NatlMedJIndia_2020_33_2_89_310984_t4.jpg){kind=link}

When to suspect
ABCDE approach. Patients can have multiple naevi, which may remain stable over time. Not all lesions require evaluation for suspected melanoma. However, any lesion showing the below mentioned characteristics should be considered suspicious. These are:
- Asymmetry: if a lesion is bisected, one half is not identical to the other half
- Border irregularities
- Colour variegation with presence of multiple shades of red, blue, black, grey or white
- Diameter ≥6 mm
- Evolution: a lesion that is changing in size, shape or colour, or a new lesion (most important).[13]
Ugly duckling approach. A naevus that is different from other naevi in an individual with multiple naevi should be considered suspicious even if it does not fulfil all the ABCDE criteria.[14]
Glasgow 7-point checklist. Weighted 7-point checklist for early detection of melanoma has been shown to be more sensitive than the ABCDE criteria in clinical practice.[15]
Major features (2 points for each)
- Change in size of lesion
- Irregular pigmentation
- Irregular border
Minor features (1 point for each)
- Inflammation
- Itch
- Diameter >7 mm
- Oozing or crusting of lesion
Any lesion with a score of ≥3 should be referred to a dermatologist for evaluation.
How to confirm?
A complete full-thickness excisional biopsy of suspicious lesions with 1–3 mm margin of normal skin and part of the subcutaneous fat should be performed whenever possible. Partial incisional biopsy may be acceptable if the excision of the entire lesion is not feasible.[16]
Staging work-up [Table - 2]
![[Table - 2]](#tbl_NatlMedJIndia_2020_33_2_89_310984_t5.jpg){kind=link}

Work-up for metastasis is not indicated in stages I and II melanoma with no clinical symptoms. In patients with stage III melanoma with occult lymph node metastasis, the role of metastatic work-up is controversial. Various studies have reported detection of distant metastasis in 3%–4% of such patients.[17],[18],[19] However, detection of metastasis has a major impact on the intent of therapy; hence, we suggest complete work-up for all such patients. For patients who have clinically palpable lymph nodes or evidence of metastasis, imaging is indicated to document sites of disease. These include magnetic resonance imaging (MRI) of the brain with contrast and fluorodeoxyglucose-positron emission tomography-computed tomography (PET-CT) of the whole body; if unavailable contrast-enhanced CT of the chest, abdomen and pelvis can be done. Although PET-CT can be false- positive and -negative, it is rapidly replacing other modalities for metastatic work-up. In a meta-analysis the median sensitivity of PET-CT for staging was 86% and specificity was 91%, whereas those for CT only were 63% and 78%, respectively.[20]
Prognostic Factors
First described by Clark et al.[21] and Breslow et al.,[22] the depth of invasion of the primary tumour has been the most important prognostic factor in localized cutaneous melanoma, in traditional staging systems.[23] In a large multicentre analysis of over 2000 patients with melanoma, it was found that Breslow thickness, mitotic rate, ulceration and sentinel lymph node status were associated with survival.[24] Tumour node metastasis (TNM) stage has important prognostic implications and co-relates directly with survival.[25] Other important prognostic factors include advanced age, sex (females have a better prognosis), anatomical location (upper extremity and face have a better prognosis than head and neck, trunk and lower extremity), growth pattern (nodular has a worse prognosis than superficial spreading), extracapsular nodal extension, pre-existing naevi (better prognosis), margins of resection, site and size of metastasis (skin only have a better prognosis than visceral metastasis) and lymphovascular invasion.[26],[27],[28],[29],[30] Elevated lactate dehydrogenase (LDH) is an important marker for poor prognosis and has been shown, in a meta-analysis, to be a predictive marker of benefit from immunotherapy and targeted therapies.[31]
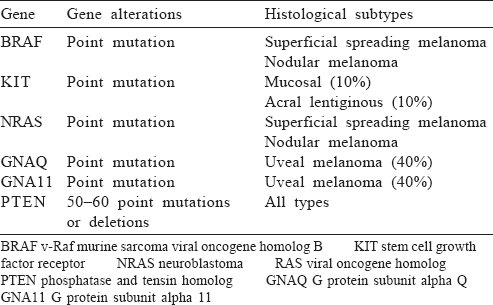
Mutation Testing in Melanoma
Mutations in the BRAF gene leading to MAP kinase pathway activation are the most common driver mutations seen in 40%–60% of all cases of melanoma.[32] The mutations most often seen in the BRAF gene are V600E (70%–80%) and V600K (20%).[32] BRAF is also the most common targetable mutation in metastatic melanoma. Real-time reverse transcription quantitative polymerase chain reaction (RT-qPCR) is the method of choice for biomarker testing. For BRAF, both V600E and V600K should be tested as both are sensitive to BRAF inhibitors.
Other important targetable mutation in melanoma is c-KIT, which is seen in 15%–40% of cases of acral melanoma, mucosal melanoma and melanoma arising from chronically sun-damaged skin but is rare in other types.[33] Since acral melanoma is more common in Asian countries, especially India, c-KIT is an important target for our population. For c-KIT mutated melanoma, mutations in exon 11 are more common than exon 9, 13, 17 and 18.[34],[35] Preferential testing for exon 11 and 9 should be done if tissue quantity is the limiting factor. Either direct sequencing or RT-qPCR can be used with the latter being the preferred method. Excluding c-KIT mutations based on immunohistochemistry for CD117 is unreliable and should not be done.[36]
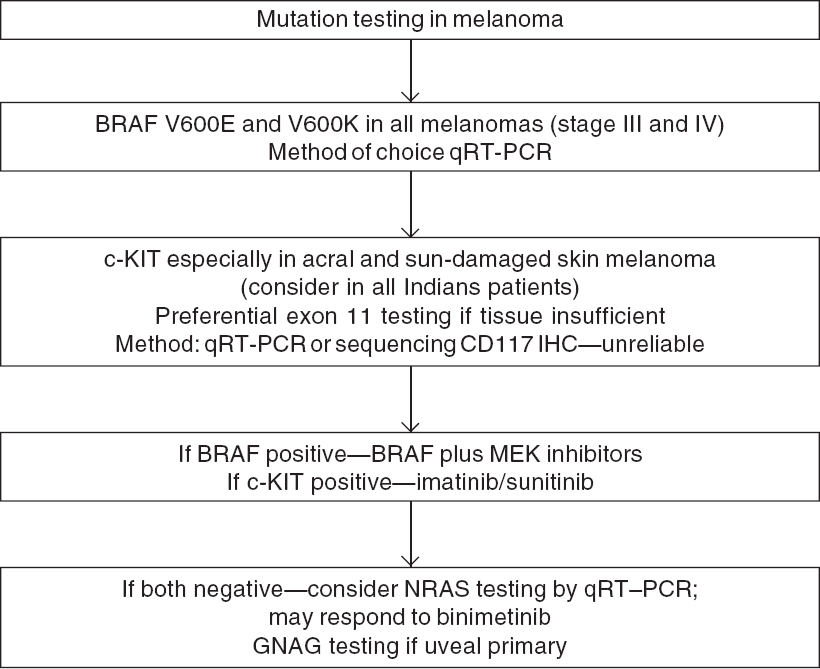
At present, guidelines recommend testing for BRAF mutation in all advanced melanoma and c-KIT testing for acral and mucosal melanomas.[37] If BRAF mutation is negative, further molecular testing for NRAS, GNA11 or GNAQ (uveal primary) can be considered as patients with NRAS mutations have responded to MEK inhibitors in phase 2 trials.[38] An algorithm for mutation testing is presented in [Figure - 1] and key genetic alterations are given in [Table - 3].
![[Figure - 1]](#fig_NatlMedJIndia_2020_33_2_89_310984_f1.jpg){kind=link}
![[Table - 3]](#tbl_NatlMedJIndia_2020_33_2_89_310984_t6.jpg){kind=link}
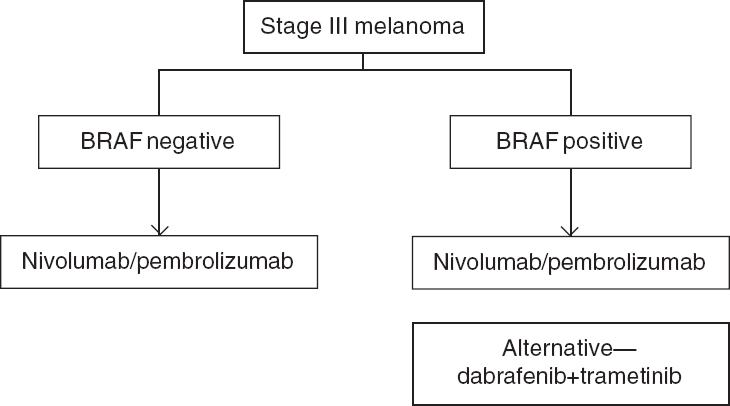 |
| Figure 1: Mutation testing algorithm in melanoma |
Adjuvant Therapy in Resected Melanoma
Indications for adjuvant therapy
All patients with localized melanoma should undergo wide local excision. Management of occult lymph nodes is controversial and the role of extensive surgery in the era of adjuvant immunotherapy is not well defined. While regional lymph- adenectomy is standard for clinically positive nodes, the role of completion surgery in sentinel lymph node biopsy (SLNB) positive patients is controversial with many randomized trials not showing survival benefit.[39],[40] However, SLNB should be done for clinically negative nodes as it provides prognostic information and is necessary for entry into clinical trials for adjuvant therapy.[41]
At present, all patients with lymph nodal involvement (including satellite/in-transit metastasis) warrant adjuvant immune/targeted therapy after surgery including those who have clinically negative lymph nodes but positive SLNB, irrespective of the size of the deposit. The best agents, toxicities and optimal duration of therapy are described in [Table - 4].
![[Table - 4]](#tbl_NatlMedJIndia_2020_33_2_89_310984_t7.jpg){kind=link}

Immunotherapy
Ipilimumab. Ipilimumab is an anti-CTLA-4 antibody which improved recurrence-free survival (RFS), metastasis-free survival and OS in phase 3 EORTC 18 071 trial in the adjuvant setting compared to placebo; the dose was 10 mg/kg and the duration of therapy was 3 years.[42],[43] However, 54% of patients experienced grades 3–4 adverse effects with treatment discontinuation rates of 41.6%. Five patients (1.1%) died during treatment. Notably, the accepted dose of ipilimumab in metastatic setting is 3 mg/kg,[44] and hence the higher dose used in this trial could explain the higher toxicity.[42] Ipilimumab has been replaced by other immuno-oncology drugs in the adjuvant setting due to its unacceptable toxicity profile.
Nivolumab. Nivolumab is an anti-programmed cell death protein 1 (PD-1) inhibitor which improved RFS and OS in resected melanoma compared with ipilimumab in CheckMate 238 trial (hazard ratio [HR] for disease recurrence or death, 0.66; 95% CI 0.54–0.81) at a dose of 3 mg/kg every 2 weeks for 1 year; grades 3–4 toxicity was 14.4% in nivolumab compared to 45.9% in ipilimumab with no treatment-related deaths; treatment discontinuation was 4% with nivolumab compared to 30% with ipilimumab; and all major pre-specified subgroups had same degree of benefit.[45],[46] Approximately 42% of patients in this trial had BRAF mutation, demonstrating efficacy of nivolumab in this subset. This trial established nivolumab at 3 mg/kg for 1 year as standard adjuvant therapy for resected stage III melanoma.
Pembrolizumab. Pembrolizumab is also an anti PD-1 antibody similar to nivolumab. It was approved for adjuvant therapy in resected stage III melanoma based on KEYNOTE-054 trial in which pembrolizumab at 200 mg thrice weekly for 18 doses significantly improved RFS compared to placebo (HR 0.57, 98.4% CI 0.43–0.74).[47] As in CheckMate 238 trial of nivolumab, PD-L1 positivity was not a predictor of response. Adverse event profile was similar to nivolumab.
BRAF inhibitors in adjuvant setting
BRAF mutation is the most common driver mutation in advanced melanoma.[32] As in metastatic melanoma, combinations of BRAF and MEK inhibitors have been tested in adjuvant setting. In the phase 3 randomized trial Combi-AD,[48],[49] dabrafenib and trametinib for 1 year improved RFS and OS with an estimated 54% cure rate (HR 0.49 for RFS and 0.57 for OS); treatment benefits were observed irrespective of baseline factors. Grade 3–4 adverse events occurred in 41% of patients, the most common being fatigue and pyrexia; the rate of new squamous cell skin cancers was similar between the two arms. Twenty-six per cent patients discontinued trial drug. Another phase 3 trial of BRAF inhibitor vemurafenib failed to meet its primary end-point of disease-free survival.[50] If data from immunotherapy trials and combi-AD are compared, 2-year RFS rates are around 60% in both, with higher toxicity in the combi-AD trial compared to nivolumab/ pembrolizumab. However, the combination seems to be better tolerated than ipilimumab with benefit of adjuvant therapy extending to all subgroups including stage IIIA melanoma.
Which agent to choose?
Given the evidence, both nivolumab and pembrolizumab are first-line options for adjuvant treatment irrespective of PDL-1 and BRAF mutation status. In our view, ipilimumab can be considered a second-line option for patients who progress on nivolumab/pembrolizumab. However, its major toxicity and dose consideration need to be kept in mind. BRAF plus MEK inhibitors remain an option in BRAF mutant patients keeping in mind their adverse event profile. The optimal duration of therapy is 1 year. Patients with stage IIIA melanoma especially with sentinel node deposit <1 mm have a 91% 5-year RFS and observation can be considered a valid treatment option for this subset of patients.[51] The benefit of using nivolumab/ pembrolizumab after progression on either agent is a pertinent research question [Figure - 2]
![[Figure - 2]](#fig_NatlMedJIndia_2020_33_2_89_310984_f2.jpg){kind=link}
 |
| Figure 2: Adjuvant therapy in stage III melanoma |
Metastatic Melanoma: Where Do We Stand?
Immunotherapy in metastatic melanoma
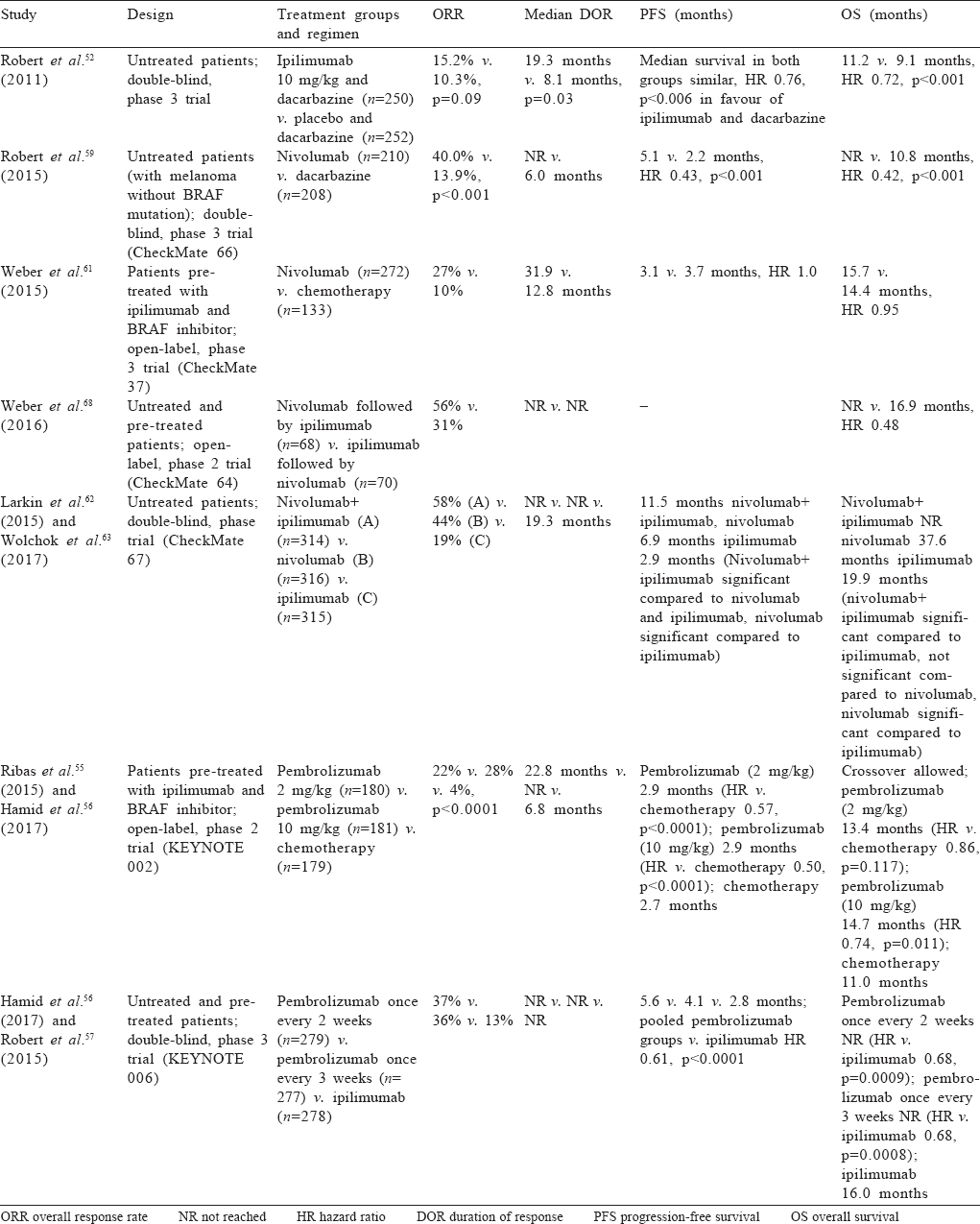
Ipilimumab. Ipilimumab in combination with dacarbazine improved OS in treatment naäve patients compared to dacarbazine alone with 5-year survival approaching 20% with ipilimumab.[52],[53] The dose used in this trial was 10 mg/kg and resulted in 56% of patients having grades 3–4 adverse events. The responses obtained with ipilimumab were durable with a plateau in survival curve after 3 years.[54] Another randomized trial which compared a dose of 10 mg/kg versus 3 mg/kg in unresectable/metastatic setting had improved survival with higher dose albeit at much higher toxicity.[44]
Pembrolizumab
Ipilimumab refractory disease. Pembrolizumab at doses of 2 mg/kg/3 weekly, 10 mg/kg/3 weekly improved progression-free survival (PFS) compared to chemotherapy in ipilimumab refractory patients (KEYNOTE-002 trial) with significantly less toxicity.[55] Both pembrolizumab arms had similar outcomes. The trial showed only a trend towards superior OS likely due to heavy crossover.[56]
Immunotherapy naïve patients. KEYNOTE-006 phase 3 trial compared two doses of pembrolizumab (10 mg/kg/2 weekly and 10 mg/kg/3 weekly) versus ipilimumab (3 mg/kg/3 weeks for 4 doses). Pembrolizumab was given for a fixed interval of 2 years. Around 35% of these patients were BRAF-positive and had received prior targeted therapy. Pembrolizumab improved response rates (42% v. 17%), PFS (2 year: 30% v. 14%) and OS (4 year: 41% v. 34%) with no difference between the two pembrolizumab arms.[57],[58] Grades 3–4 adverse events were reported in 14%–17% of patients in the pembrolizumab group and 20% of patients in the ipilimumab group. Patterns of toxicity differed with colitis being more common with ipilimumab (7%–8%; grades 3–4) whereas thyroid dysfunction (10%) and pneumonitis (2%) were more common with pembrolizumab.
Nivolumab. Initial phase 3 trials with nivolumab in treatment-naïve patients without BRAF mutation provided evidence of its superiority over chemotherapy (median OS 38 v. 11 months, overall response rate 40% v. 14% and PFS 5.1 v. 2.2 months; CheckMate 066 trial).[59],[60] In patients who had received prior ipilimumab, nivolumab compared with chemotherapy improved response rates and duration of response. However, in this trial there was no survival difference which can be due to poor compliance in the chemotherapy arm and severe imbalance of poor prognostic factors in the nivolumab arm which had more patients with brain metastasis and elevated LDH. Based on these data, the US Food and Drug Administration approved nivolumab for metastatic melanoma at 3 mg/kg/2 weekly;[61] later a flat dose of 240 mg/2 weekly or 480 mg/4 weekly was also approved. The adverse event profile of nivolumab was similar to that seen in the adjuvant setting.
Combination immunotherapy. To further improve outcomes, combination immunotherapy was tested in the landmark phase 3 trial—CheckMate 067.[62] In this trial, a combination of nivolumab and ipilimumab (induction for 4 cycles, nivolumab dose 1 mg/ kg and ipilimumab dose 3 mg/kg) was tested against nivolumab single agent (3 mg/kg/2 weekly) and ipilimumab single agent (3 mg/kg/2 weekly 4 doses) with maintenance nivolumab in the first two arms. The combination and single agent nivolumab arm did better in all end-points including response rates (58%, 44% and 19%), PFS and OS compared to ipilimumab. Three-year PFS was 39%, 32% and 10%, respectively. Three-year OS was 58%, 52% and 34% (HR for the combination v. ipilimumab 0.55, 95% CI 0.45–0.69, and HR for nivolumab alone v. ipilimumab 0.65, 95% CI 0.53–0.80). Although PFS was better with combination than with single agent nivolumab, it was marginal and the trial was not adequately powered to detect this difference.[63] Compared to patients without BRAF mutation, those with mutation had better PFS and OS than patients with combination immunotherapy providing a statistically significant PFS benefit over single agent nivolumab in this subset. Around 59% of patients receiving combination therapy had grades 3–4 adverse events compared to 21% with nivolumab alone and 28% with ipilimumab alone. Treatment had to be discontinued in 39% in the combination arm due to toxicity compared to 12% in the nivolumab arm. The lower dose of the combination was tested in the CheckMate 511 trial (ipilimumab (1 mg/kg) with nivolumab at 3 mg/kg); although grades 3–4 adverse events decreased (34% in this trial v. 48% in the original trial), they were still significant and the trial was not adequately powered to ascertain non-inferiority.[64]
Stopping immunotherapy?
In the KEYNOTE 006 trial the duration of therapy was fixed at 2 years and it was observed that around 20% of patients who completed planned pembrolizumab treatment discontinued therapy after 2 years and 86% of those remained disease-free on long-term follow-up (median 20 months) suggesting a potential cure and durable response.[65],[66] This trial also highlighted that the initial response to immunotherapy can be a surrogate for survival as 28% of patients who had complete response with pembrolizumab had PFS rate of 96% at 18 months. For patients who progressed, around 50% responded to a re-challenge suggesting that the initial good response can be used as a pointer to subsequent response on progression. A similar phase 1B KEYNOTE 001 trial showed feasibility of stopping pembrolizumab after a minimum of 6 months or 2 cycles after CR whichever was later.[67] Tumour size, PD-L1 status and LDH were predictors of response to immunotherapy; however, their validation requires larger trials. Similar trials for nivolumab and combination immunotherapy are under way. Based on this data, 2 years of immunotherapy may be considered standard in responding patients.
What is the best frontline therapy?
Based on the above data, although combination immunotherapy provides better response rates and PFS rates compared to single agent immunotherapy, single agent nivolumab and pembrolizumab fare well in terms of all end-points with no significant difference in OS compared to a combination with a favourable side-effect profile. Effectiveness is maintained in BRAF mutated tumours also. In our setting where cost is a major issue, single agent nivolumab is probably the best frontline option. As shown for pembrolizumab in the KEYNOTE 006 trial, patients with CR or those who have maintained response for 2 years can discontinue therapy without losing effectiveness. Though this approach has not been tested for nivolumab, conceptually it should be valid. Best treatment after progression on first-line immunotherapy with nivolumab and pembrolizumab is uncertain; combination immunotherapy or single agent ipilimumab can be considered in this setting. Better response rates and OS have been demonstrated in nivolumab to ipilimumab sequence than reverse but toxicity was similar to that for the combination.[68] A comparison of various trials of immunotherapy in metastatic disease is summarized in [Table - 5].
![[Table - 5]](#tbl_NatlMedJIndia_2020_33_2_89_310984_t8.jpg){kind=link}
Immune-related adverse events (irAEs)
irAEs are a unique spectrum of side-effects associated with immune checkpoint inhibition. In various clinical trials, ipilimumab was consistently more toxic than nivolumab/ pembrolizumab with grades 3–4 adverse events seen in 50% of patients with ipilimumab and 15% with nivolumab/ pembrolizumab. irAEs require strict monitoring and are treatable with steroids if detected early. The spectrum of irAEs seen with anti-CTLA-4 and anti-PD-1 antibodies is different. Endocrino-pathies (thyroiditis, hypophysitis, adrenal insufficiency) and colitis are more frequent with anti-CTLA-4 and pneumonitis and hepatitis seen more commonly with anti-PD-1. Appearance of irAEs is unpredictable and does not corelate with the cumulative dose.[69]
Management of irAEs depends on the grade of the adverse event. Temporary discontinuation of immunotherapy is required for a grade 2 event, steroids may be added at 0.5 mg/kg if symptoms do not resolve within a week. The drug should be restarted once symptoms are grade 1 or less. Immunotherapy should be permanently discontinued for grades 3–4 adverse events and management requires high dose steroids (1–2 mg/ kg) with refractory cases requiring infliximab. Steroids can be tapered slowly once symptoms decrease to grade 1 or less.[69]
BRAF and MEK inhibitors: Two better than one?
Concept of dual inhibition. Targeted therapy against BRAF and MEK is an important treatment option for patients with BRAF/NRAS mutated tumours. There are no data from randomized trials comparing BRAF/MEK inhibitors with immunotherapy in BRAF mutated melanoma and optimal sequencing is unknown.
Both dabrafenib and vemurafenib improved response rates, PFS and OS in pivotal phase 3 trials when compared with dacarbazine.[70],[71],[72],[73] However, single agent BRAF inhibitors caused keratoacanthomas and new squamous cell carcinomas in 20%–25% of patients[74] and new melanomas in around 2%. The accepted hypothesis implicates BRAF independent downstream activation of MAP kinase pathway as a possible mechanism for this toxicity and that a second step inhibition with MEK inhibitors might circumvent this complication. Based on the above hypothesis, combinations of BRAF inhibitors with MEK inhibitors have been tested in melanoma.
Supporting Evidence
The first combination tried was dabrafenib with trametinib in two randomized trials. In the Combi-AD study, a combination of dabrafenib and trametinib was compared to dabrafenib alone; the combination arm showed unprecedented response rates (68% v. 55%), median PFS (11 v. 8.8 months, HR 0.67, 95% CI 0.53–0.84) and OS (median 25.1 v. 18.7 months, HR 0.71, 95% CI 0.55–0.92). Around 20% of patients treated with the combination maintained disease control on long-term follow-up compared to 6% in dabrafenib alone.[75],[76] Cutaneous toxicity was decreased with the combination with squamous cell carcinoma seen in 9% with dabrafenib alone and 3% with the combination. Pyrexia, chills and gastrointestinal toxicity was more in the combination leading to higher discontinuation rates (11% v. 7%). Similar results were seen when the above combination was compared with vemurafenib in a phase 3 trial.[77] These trials showed a median PFS of 11 months and median OS of 25 months; patients having less than three sites of metastasis and normal LDH had significantly better outcomes.[78]
A combination of vemurafenib and cobimetinib tested in a phase 3 trial showed similar results to dabrafenib and trametinib with similar toxicity.[79]
In the phase 3 COLUMBUS trial,[80],[81] a combination of encorafenib and binimetinib showed prolonged PFS and OS compared to vemurafenib alone or encorafenib alone (PFS 14.3 v. 9.7 months, OS 33.6 v. 23.5 months). Numerically this trial had the longest median PFS and OS, but none of these combinations have been directly compared and cross-trial comparisons should be interpreted cautiously. A combination of dabrafenib and trametinib has shown great efficacy in brain metastasis with response rates of 55%. Binimetinib has also shown activity in NRAS mutated tumours and can be used in this subset of patients; however, it is not currently approved for this indication.[82]
When To Use?
To summarize, a combination of BRAF and MEK inhibitors is an important treatment option in BRAF mutant melanoma with best results so far for the encorafenib and binimetinib combination. However, there are no direct comparisons between various BRAF and MEK inhibitors and between these targeted therapy and immunotherapy. Moreover, pembrolizumab showed excellent response and survival in BRAF mutated tumours, making it a reasonable first-line choice in this subset. Also, BRAF and MEK inhibitors, although approved by DCGI, they are not currently available in India making targeted therapy a second-line option in BRAF mutated melanoma [Figure - 3].
![[Figure - 3]](#fig_NatlMedJIndia_2020_33_2_89_310984_f3.jpg){kind=link}
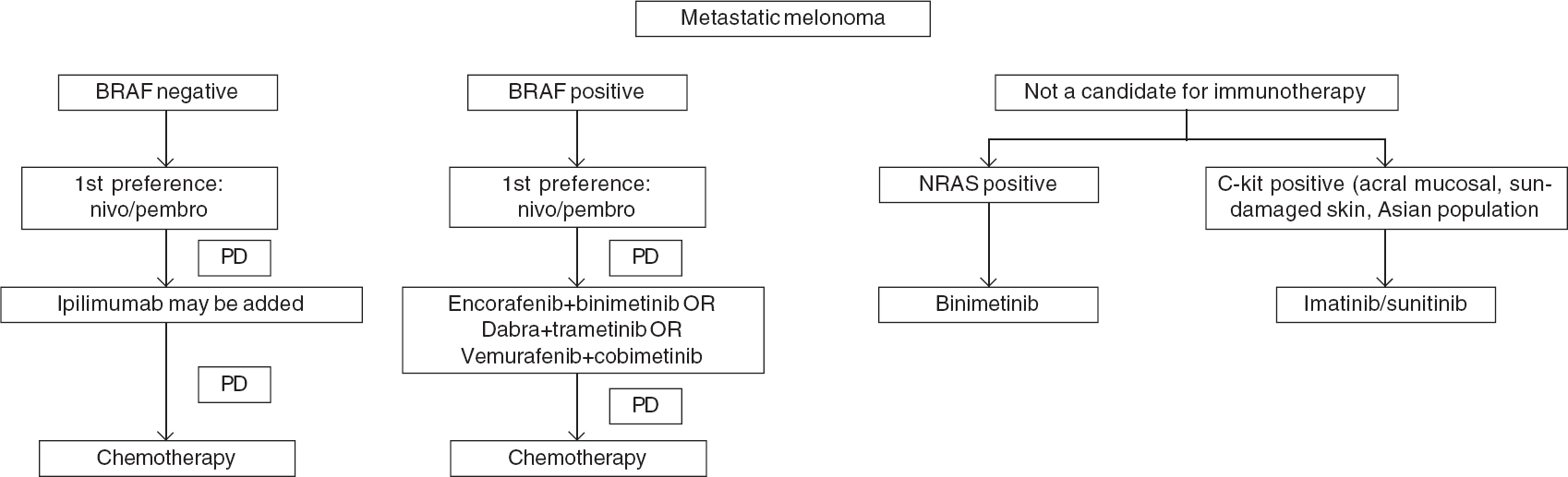 |
| Figure 3: Management algorithm for metastatic melanoma |
C-KIT and Imatinib: Value in The Indian Setting
Mutations in c-KIT are more often seen in acral and mucosal melanomas; since acral melanoma is more common in India compared to the West, targeting c-KIT with tyrosine kinase inhibitors is a valid treatment option. Three phase 2 trials have tested imatinib in c-KIT mutant melanoma; results are variable with partial responses seen in 23%–54% of patients.[34],[83],[84] Interestingly, only patients with exon 11 (L576P) and exon 13 (K642E) responded; patients with KIT amplification without mutation did not respond. Similar activity has been observed with nilotinib in previously treated patients with imatinib and treatment-naäve patients,[85],[86] dasatanib,[87] sorafenib[88] and sunitinib.[89]
Does Chemotherapy Have A Role in The Current Scenario?
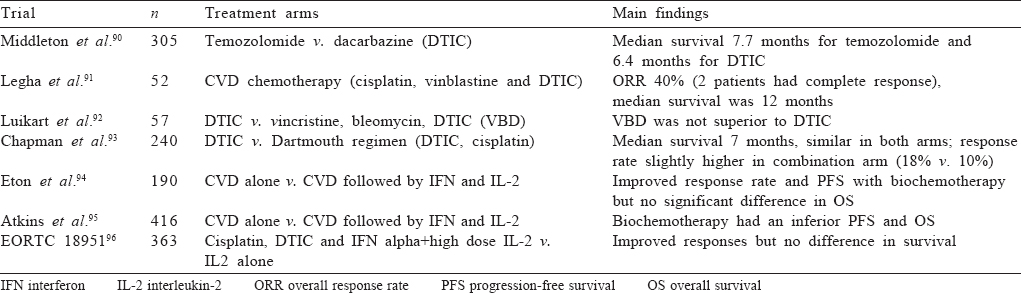
Chemotherapy in metastatic melanoma has been disappointing. A number of agents have been tried alone and in combination with disappointing results. Dacarbazine has been the traditional comparator arm for most immunotherapy trials and is considered the most active chemotherapeutic drug in melanoma. Other drugs such as temozolomide, platinum compounds and taxanes have been tested without much benefit. Addition of interleukin 2 has not shown survival benefit in any trial. In the present scenario, chemotherapy may be considered for patients who are not eligible or have progressed on immunotherapy and are not eligible for clinical trials as a last resort. The major chemotherapy trials are summarized in [Table - 6].
![[Table - 6]](#tbl_NatlMedJIndia_2020_33_2_89_310984_t9.jpg){kind=link}
Future Perspectives
Patients with high-risk early-stage disease (>4 mm thickness without ulceration and >2 mm thickness with ulceration but negative nodes) have higher relapse risk, but they have not been evaluated in phase 3 trials involving adjuvant immunotherapy or targeted therapy. Trials in this population is an unmet need but they do not merit adjuvant therapy outside of a trial setting at present. Adequately powered trials comparing combination immunotherapy with single agent nivolumab are required to answer the question of best frontline option in metastatic melanoma. Since no direct comparisons of immunotherapy and targeted therapy are available, trials to answer that question in BRAF mutated melanoma are also anticipated. Although preliminary evidence indicates that immunotherapy can be discontinued after 2 years in metastatic melanoma, concrete evidence to this effect is lacking.
To conclude, based on the results with the present-day immunotherapy and targeted therapy in melanoma, it appears that the intent of treatment in metastatic setting may be considered as curative as more than half the patients have long-term survival with many patients able to discontinue treatment without losing response.
Conflicts of interest. Nil
| 1. | Ganesan P, Bakhshi S. Systemic therapy for melanoma. Natl Med J India 2010;23:21–7. [Google Scholar] |
| 2. | Garbe C, Eigentler TK, Keilholz U, Hauschild A, Kirkwood JM. Systematic review of medical treatment in melanoma: Current status and future prospects. Oncologist 2011;16:5–24. [Google Scholar] |
| 3. | Guy GP Jr, Thomas CC, Thompson T, Watson M, Massetti GM, Richardson LC, et al. Vital signs: Melanoma incidence and mortality trends and projections––United States, 1982–2030. MMWR Morb Mortal Wkly Rep 2015;64:591–6. [Google Scholar] |
| 4. | Ferlay J, Colombet M, Soerjomataram I, Mathers C, Parkin DM, Piñeros M, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer 2019;144:1941–53. [Google Scholar] |
| 5. | Parkin DM, Pisani P, Ferlay J. Estimates of the worldwide incidence of 25 major cancers in 1990. Int J Cancer 1999;80:827–41. [Google Scholar] |
| 6. | Aitken JF, Youlden DR, Baade PD, Soyer HP, Green AC, Smithers BM. Generational shift in melanoma incidence and mortality in Queensland, Australia, 1995–2014. Int J Cancer 2018;142:1528–35. [Google Scholar] |
| 7. | Pandey M, Mathew A, Abraham EK, Ahamed IM, Nair KM. Primary malignant melanoma of the mucous membranes. Eur J Surg Oncol 1998;24:303–7. [Google Scholar] |
| 8. | Cormier JN, Xing Y, Ding M, Lee JE, Mansfield PF, Gershenwald JE, et al. Ethnic differences among patients with cutaneous melanoma. Arch Intern Med 2006; 166:1907–14. [Google Scholar] |
| 9. | Elwood JM, Jopson J. Melanoma and sun exposure: An overview of published studies. Int J Cancer 1997;73:198–203. [Google Scholar] |
| 10. | Wu S, Han J, Laden F, Qureshi AA. Long-term ultraviolet flux, other potential risk factors, and skin cancer risk: A cohort study. Cancer Epidemiol Biomarkers Prev 2014;23:1080–9. [Google Scholar] |
| 11. | Harland M, Goldstein AM, Kukalizch K, Taylor C, Hogg D, Puig S, et al. A comparison of CDKN2A mutation detection within the Melanoma Genetics Consortium (GenoMEL). Eur J Cancer 2008;44:1269–74. [Google Scholar] |
| 12. | Ward WH, Lambreton F, Goel N, Yu JQ, Farma JM. Clinical presentation and staging of melanoma. In: Ward WH, Farma JM (eds). Cutaneous melanoma: Etiology and therapy. Brisbane (AU):Codon Publications; 2017:79–89. [Google Scholar] |
| 13. | Abbasi NR, Shaw HM, Rigel DS, Friedman RJ, McCarthy WH, Osman I, et al. Early diagnosis of cutaneous melanoma: Revisiting the ABCD criteria. JAMA 2004; 292:2771–6. [Google Scholar] |
| 14. | Grob JJ, Bonerandi JJ. The ‘ugly duckling’ sign: Identification of the common characteristics of nevi in an individual as a basis for melanoma screening. Arch Dermatol 1998;134:103–4. [Google Scholar] |
| 15. | Walter FM, Prevost AT, Vasconcelos J, Hall PN, Burrows NP, Morris HC, et al. Using the 7-point checklist as a diagnostic aid for pigmented skin lesions in general practice: A diagnostic validation study. Br J Gen Pract 2013;63:e345–e353. [Google Scholar] |
| 16. | Marsden JR, Newton-Bishop JA, Burrows L, Cook M, Corrie PG, Cox NH, et al. Revised UK guidelines for the management of cutaneous melanoma 2010. Br J Dermatol 2010;163:238–56. [Google Scholar] |
| 17. | Miranda EP, Gertner M, Wall J, Grace E, Kashani-Sabet M, Allen R, et al. Routine imaging of asymptomatic melanoma patients with metastasis to sentinel lymph nodes rarely identifies systemic disease. Arch Surg 2004;139:831–6. [Google Scholar] |
| 18. | Gold JS, Jaques DP, Busam KJ, Brady MS, Coit DG. Yield and predictors of radiologic studies for identifying distant metastases in melanoma patients with a positive sentinel lymph node biopsy. Ann Surg Oncol 2007;14:2133–40. [Google Scholar] |
| 19. | Aloia TA, Gershenwald JE, Andtbacka RH, Johnson MM, Schacherer CW, Ng CS, et al. Utility of computed tomography and magnetic resonance imaging staging before completion lymphadenectomy in patients with sentinel lymph node-positive melanoma. J Clin Oncol 2006;24:2858–65. [Google Scholar] |
| 20. | Xing Y, Bronstein Y, Ross MI, Askew RL, Lee JE, Gershenwald JE, et al. Contemporary diagnostic imaging modalities for the staging and surveillance of melanoma patients: A meta-analysis. J Natl Cancer Inst 2011;103:129–42. [Google Scholar] |
| 21. | Clark WH Jr, From L, Bernardino EA, Mihm MC. The histogenesis and biologic behavior of primary human malignant melanomas of the skin. Cancer Res 1969; 29:705–27. [Google Scholar] |
| 22. | Breslow A. Thickness, cross-sectional areas and depth of invasion in the prognosis of cutaneous melanoma. Ann Surg 1970;172:902–8. [Google Scholar] |
| 23. | Green AC, Baade P, Coory M, Aitken JF, Smithers M. Population-based 20-year survival among people diagnosed with thin melanomas in Queensland, Australia. J Clin Oncol 2012;30:1462–7. [Google Scholar] |
| 24. | Maurichi A, Miceli R, Camerini T, Mariani L, Patuzzo R, Ruggeri R, et al. Prediction of survival in patients with thin melanoma: Results from a multiinstitution study. J Clin Oncol 2014;32:2479–85. [Google Scholar] |
| 25. | Gershenwald JE, Scolyer RA, Hess KR, et al. Melanoma of the skin. In: Amin MB, Edge S, Greene F, Byrd DR, Brookland RK, Washington MK, et al. (eds). AJCC Cancer Staging Manual. 8th ed. Chicago:American Joint Committee on Cancer; 2017:563. [Google Scholar] |
| 26. | Thompson JF, Soong SJ, Balch CM, Gershenwald JE, Ding S, Coit DG, et al. Prognostic significance of mitotic rate in localized primary cutaneous melanoma: An analysis of patients in the multi-institutional American Joint Committee on Cancer melanoma staging database. J Clin Oncol 2011;29:2199–205. [Google Scholar] |
| 27. | Scoggins CR, Ross MI, Reintgen DS, Noyes RD, Goydos JS, Beitsch PD, et al. Gender-related differences in outcome for melanoma patients. Ann Surg 2006;243:693–8. [Google Scholar] |
| 28. | Callender GG, Egger ME, Burton AL, Scoggins CR, Ross MI, Stromberg AJ, et al. Prognostic implications of anatomic location of primary cutaneous melanoma of 1 mm or thicker. Am J Surg 2011;202:659–64. [Google Scholar] |
| 29. | Balch CM, Soong SJ, Gershenwald JE, Thompson JF, Reintgen DS, Cascinelli N, et al. Prognostic factors analysis of 17,600 melanoma patients: Validation of the American Joint Committee on Cancer melanoma staging system. J Clin Oncol 2001;19:3622–34. [Google Scholar] |
| 30. | Lattanzi M, Lee Y, Simpson D, Moran U, Darvishian F, Kim RH, et al. Primary melanoma histologic subtype: Impact on survival and response to therapy. J Natl Cancer Inst 2019;111:180–8. [Google Scholar] |
| 31. | Petrelli F, Ardito R, Merelli B, Lonati V, Cabiddu M, Seghezzi S, et al. Prognostic and predictive role of elevated lactate dehydrogenase in patients with melanoma treated with immunotherapy and BRAF inhibitors: A systematic review and meta- analysis. Melanoma Res 2019;29:1–2. [Google Scholar] |
| 32. | Omholt K, Platz A, Kanter L, Ringborg U, Hansson J. NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clin Cancer Res 2003;9:6483–8. [Google Scholar] |
| 33. | Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol 2006;24:4340–6. [Google Scholar] |
| 34. | Carvajal RD, Antonescu CR, Wolchok JD, Chapman PB, Roman RA, Teitcher J, et al. KIT as a therapeutic target in metastatic melanoma. JAMA 2011;305:2327–34. [Google Scholar] |
| 35. | Beadling C, Jacobson-Dunlop E, Hodi FS, Le C, Warrick A, Patterson J, et al. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res 2008;14:6821–8. [Google Scholar] |
| 36. | Abu-Abed S, Pennell N, Petrella T, Wright F, Seth A, Hanna W. KIT gene mutations and patterns of protein expression in mucosal and acral melanoma. J Cutaneous Med Surg 2012;16:135–42. [Google Scholar] |
| 37. | Martín-Algarra S, Fernández-Figueras MT, López-Martín JA, Santos-Briz A, Arance A, Lozano MD, et al. Guidelines for biomarker testing in metastatic melanoma: A National Consensus of the Spanish Society of Pathology and the Spanish Society of Medical Oncology. Clin Transl Oncol 2014;16:362–73. [Google Scholar] |
| 38. | Dummer R, Hauschild A, Lindenblatt N, Pentheroudakis G, Keilholz U; On behalf of the ESMO Guidelines Committee. Cutaneous melanoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2015;26 (5 Suppl):v126–v132. [Google Scholar] |
| 39. | Faries MB, Thompson JF, Cochran AJ, Andtbacka RH, Mozzillo N, Zager JS, et al. Completion dissection or observation for sentinel-node metastasis in melanoma. N Engl J Med 2017;376:2211–22. [Google Scholar] |
| 40. | Leiter U, Stadler R, Mauch C, Hohenberger W, Brockmeyer N, Berking C, et al. Complete lymph node dissection versus no dissection in patients with sentinel lymph node biopsy positive melanoma (DeCOG-SLT): A multicentre, randomised, phase 3 trial. Lancet Oncol 2016;17:757–67. [Google Scholar] |
| 41. | Morton DL, Thompson JF, Cochran AJ, Mozzillo N, Elashoff R, Essner R, et al. Sentinel-node biopsy or nodal observation in melanoma. N Engl J Med 2006;355:1307–17. Erratum in: N Engl J Med 2006;355:1944. [Google Scholar] |
| 42. | Eggermont AM, Chiarion-Sileni V, Grob JJ, Dummer R, Wolchok JD, Schmidt H, et al. Adjuvant ipilimumab versus placebo after complete resection of high-risk stage III melanoma (EORTC 18071): A randomised, double-blind, phase 3 trial. Lancet Oncol 2015;16:522–30. Erratum in: Lancet Oncol 2015;16:e262. Lancet Oncol 2016;17:e223. [Google Scholar] |
| 43. | Eggermont AM, Chiarion-Sileni V, Grob JJ, Dummer R, Wolchok JD, Schmidt H, et al. Prolonged survival in stage III melanoma with ipilimumab adjuvant therapy. N Engl J Med 2016;375:1845–55. [Google Scholar] |
| 44. | Ascierto PA, Del Vecchio M, Robert C, Mackiewicz A, Chiarion-Sileni V, Arance A, et al. Ipilimumab 10 mg/kg versus ipilimumab 3 mg/kg in patients with unresectable or metastatic melanoma: A randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol 2017;18:611–22. [Google Scholar] |
| 45. | Weber J, Mandala M, Del Vecchio M, Gogas HJ, Arance AM, Cowey CL, et al. Adjuvant nivolumab versus ipilimumab in resected stage iii or iv melanoma. N Engl J Med 2017;377:1824–35. [Google Scholar] |
| 46. | Weber JS, Mandalà M, Del Vecchio M, Gogas H, Arance AM, Cowey CL, et al. Adjuvant therapy with nivolumab (NIVO) versus ipilimumab (IPI) after complete resection of stage III/IV melanoma: Updated results from a phase III trial (CheckMate 238). J Clin Oncol 2018;36:18s. [Google Scholar] |
| 47. | Eggermont AM, Blank CU, Mandala M, Long GV, Atkinson V, Dalle S, et al. Adjuvant pembrolizumab versus placebo in resected stage III melanoma. N Engl J Med 2018;378:1789–801. [Google Scholar] |
| 48. | Long GV, Hauschild A, Santinami M, Atkinson V, Mandalà M, Chiarion-Sileni V, et al. Adjuvant dabrafenib plus trametinib in stage III BRAF-mutated melanoma. N Engl J Med 2017;377:1813–23. [Google Scholar] |
| 49. | Hauschild A, Dummer R, Schadendorf D, Santinami M, Atkinson V, Mandalà M, et al. Longer follow-up confirms relapse-free survival benefit with adjuvant dabrafenib plus trametinib in patients with resected BRAF V600-mutant stage III melanoma. J Clin Oncol 2018;36:3441–9. [Google Scholar] |
| 50. | Maio M, Lewis K, Demidov L, Mandalà M, Bondarenko I, Ascierto PA, et al. Adjuvant vemurafenib in resected, BRAFV600 mutation-positive melanoma (BRIM8): A randomised, double-blind, placebo-controlled, multicentre, phase 3 trial. Lancet Oncol 2018;19:510–20. [Google Scholar] |
| 51. | McKean MA, Amaria RN. Multidisciplinary treatment strategies in high-risk resectable melanoma: Role of adjuvant and neoadjuvant therapy. Cancer Treat Rev 2018;70:144–53. [Google Scholar] |
| 52. | Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011;364:2517–26. [Google Scholar] |
| 53. | Maio M, Grob JJ, Aamdal S, Bondarenko I, Robert C, Thomas L, et al. Five-year survival rates for treatment-naive patients with advanced melanoma who received ipilimumab plus dacarbazine in a phase III trial. J Clin Oncol 2015;33:1191–6. [Google Scholar] |
| 54. | Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol 2015;33:1889–94. [Google Scholar] |
| 55. | Ribas A, Puzanov I, Dummer R, Schadendorf D, Hamid O, Robert C, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): A randomised, controlled, phase 2 trial. Lancet Oncol 2015;16:908–18. [Google Scholar] |
| 56. | Hamid O, Puzanov I, Dummer R, Schachter J, Daud A, Schadendorf D, et al. Final analysis of a randomised trial comparing pembrolizumab versus investigator- choice chemotherapy for ipilimumab-refractory advanced melanoma. Eur J Cancer 2017;86:37–45. [Google Scholar] |
| 57. | Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 2015;372:2521–32. [Google Scholar] |
| 58. | Schachter J, Ribas A, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab for advanced melanoma: Final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet 2017;390:1853–62. [Google Scholar] |
| 59. | Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015;372:320–30. [Google Scholar] |
| 60. | Ascierto PA, Long GV, Robert C, Brady B, Dutriaux C, Di Giacomo AM, et al. Survival outcomes in patients with previously untreated BRAF wild-type advanced melanoma treated with nivolumab therapy: Three-year follow-up of a randomized phase 3 Trial. JAMA Oncol 2019;5:187–94. [Google Scholar] |
| 61. | Weber JS, D’Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol 2015;16:375–84. [Google Scholar] |
| 62. | Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015;373:23–34. [Google Scholar] |
| 63. | Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med 2017;377:1345–56. [Google Scholar] |
| 64. | Lebbé C, Meyer N, Mortier L, Marquez-Rodas I, Robert C, Rutkowski P, et al. Evaluation of two dosing regimens for nivolumab in combination with ipilimumab in patients with advanced melanoma: Results from the phase IIIb/IV CheckMate 511 Trial. J Clin Oncol 2019;37:867–75. [Google Scholar] |
| 65. | Robert C, Long GV, Schachter J, Mortier L, Daud A, Matteo M, et al. Long term outcomes in patients (pts) with ipilimumab (ipi)-naive advanced melanoma in the phase 3 KEYNOTE-006 study who completed pembrolizumab (pembro) treatment. J Clin Oncol 2017;35 (15 Suppl):9504. [Google Scholar] |
| 66. | Long GV, Schachter J, Ribas A, Arance AM, Grob J-J, Mortier L, et al. 4-year survival and outcomes after cessation of pembrolizumab (pembro) after 2-years in patients (pts) with ipilimumab (ipi)-naive advanced melanoma in KEYNOTE- 006. J Clin Oncol 2018;36 (15 Suppl):9503. [Google Scholar] |
| 67. | Robert C, Ribas A, Hamid O, Daud A, Wolchok JD, Joshua AM, et al. Durable complete response after discontinuation of pembrolizumab in patients with metastatic melanoma. J Clin Oncol 2018;36:1668–74. [Google Scholar] |
| 68. | Weber JS, Gibney G, Sullivan RJ, Sosman JA, Slingluff CL Jr, Lawrence DP, et al. Sequential administration of nivolumab and ipilimumab with a planned switch in patients with advanced melanoma (CheckMate 064): An open-label, randomised, phase 2 trial. Lancet Oncol 2016;17:943–55. [Google Scholar] |
| 69. | Brahmer JR, Lacchetti C, Schneider BJ, Atkins MB, Brassil KJ, Caterino JM, et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol 2018;36:1714–68. [Google Scholar] |
| 70. | Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364:2507–16. [Google Scholar] |
| 71. | McArthur GA, Chapman PB, Robert C, Larkin J, Haanen JB, Dummer R, et al. Safety and efficacy of vemurafenib in BRAF (V600E) and BRAF (V600K) mutationpositive melanoma (BRIM-3): Extended follow-up of a phase 3, randomised, open- label study. Lancet Oncol 2014;15:323–32. [Google Scholar] |
| 72. | Chapman PB, Robert C, Larkin J, Haanen JB, Ribas A, Hogg D, et al. Vemurafenib in patients with BRAFV600 mutation-positive metastatic melanoma: Final overall survival results of the randomized BRIM-3 study. Ann Oncol 2017;28:2581–7. [Google Scholar] |
| 73. | Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012;380:358–65. [Google Scholar] |
| 74. | Lacouture ME, Duvic M, Hauschild A, Prieto VG, Robert C, Schadendorf D, et al. Analysis of dermatologic events in vemurafenib-treated patients with melanoma. Oncologist 2013;18:314–22. [Google Scholar] |
| 75. | Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 2014;371:1877–88. [Google Scholar] |
| 76. | Long GV, Flaherty KT, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: Long-term survival and safety analysis of a phase 3 study. Ann Oncol 2017;28:1631–9. [Google Scholar] |
| 77. | Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 2015;372:30–9. [Google Scholar] |
| 78. | Long GV, Grob JJ, Nathan P, Ribas A, Robert C, Schadendorf D, et al. Factors predictive of response, disease progression, and overall survival after dabrafenib and trametinib combination treatment: A pooled analysis of individual patient data from randomised trials. Lancet Oncol 2016;17:1743–54. [Google Scholar] |
| 79. | Larkin J, Ascierto PA, Dréno B, Atkinson V, Liszkay G, Maio M, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 2014;371:1867–76. [Google Scholar] |
| 80. | Dummer R, Ascierto PA, Gogas HJ, Arance A, Mandala M, Liszkay G, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2018;19:603–15. [Google Scholar] |
| 81. | Dummer R, Ascierto PA, Gogas HJ, Arance A, Mandala M, Liszkay G, et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 2018;19:1315–27. [Google Scholar] |
| 82. | Dummer R, Schadendorf D, Ascierto PA, Arance A, Dutriaux C, Di Giacomo AM, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 2017;18:435–45. [Google Scholar] |
| 83. | Guo J, Si L, Kong Y, Flaherty KT, Xu X, Zhu Y, et al. Phase II, open-label, singlearm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol 2011;29:2904–9. [Google Scholar] |
| 84. | Hodi FS, Corless CL, Giobbie-Hurder A, Fletcher JA, Zhu M, Marino-Enriquez A, et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J Clin Oncol 2013;31:3182–90. [Google Scholar] |
| 85. | Carvajal RD, Lawrence DP, Weber JS, Gajewski TF, Gonzalez R, Lutzky J, et al. Phase II study of nilotinib in melanoma harboring KIT alterations following progression to prior KIT inhibition. Clin Cancer Res 2015;21:2289–96. [Google Scholar] |
| 86. | Guo J, Carvajal RD, Dummer R, Hauschild A, Daud A, Bastian BC, et al. Efficacy and safety of nilotinib in patients with KIT-mutated metastatic or inoperable melanoma: Final results from the global, single-arm, phase II TEAM trial. Ann Oncol 2017;28:1380–7. [Google Scholar] |
| 87. | Kalinsky K, Lee S, Rubin KM, Lawrence DP, Iafrarte AJ, Borger DR, et al. A phase 2 trial of dasatinib in patients with locally advanced or stage IV mucosal, acral, or vulvovaginal melanoma: A trial of the ECOG-ACRIN Cancer Research Group (E2607). Cancer 2017;123:2688–97. [Google Scholar] |
| 88. | Quintás-Cardama A, Lazar AJ, Woodman SE, Kim K, Ross M, Hwu P. Complete response of stage IV anal mucosal melanoma expressing KIT Val560Asp to the multikinase inhibitor sorafenib. Nat Clin Pract Oncol 2008;5:737–40. [Google Scholar] |
| 89. | Minor DR, Kashani-Sabet M, Garrido M, O’Day SJ, Hamid O, Bastian BC. Sunitinib therapy for melanoma patients with KIT mutations. Clin Cancer Res 2012;18: 1457–63. [Google Scholar] |
| 90. | Middleton MR, Grob JJ, Aaronson N, Fierlbeck G, Tilgen W, Seiter S, et al. Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. J Clin Oncol 2000;18:158–66. [Google Scholar] |
| 91. | Legha SS, Ring S, Papadopoulos N, Plager C, Chawla S, Benjamin R. A prospective evaluation of a triple-drug regimen containing cisplatin, vinblastine, and dacarbazine (CVD) for metastatic melanoma. Cancer 1989;64:2024–9. [Google Scholar] |
| 92. | Luikart SD, Kennealey GT, Kirkwood JM. Randomized phase III trial of vinblastine, bleomycin, and cis-dichlorodiammine-platinum versus dacarbazine in malignant melanoma. J Clin Oncol 1984;2:164–8. [Google Scholar] |
| 93. | Chapman PB, Einhorn LH, Meyers ML, Saxman S, Destro AN, Panageas KS, et al. Phase III multicenter randomized trial of the Dartmouth regimen versus dacarbazine in patients with metastatic melanoma. J Clin Oncol 1999;17:2745–51. [Google Scholar] |
| 94. | Eton O, Legha SS, Bedikian AY, Lee JJ, Buzaid AC, Hodges C, et al. Sequential biochemotherapy versus chemotherapy for metastatic melanoma: Results from a phase III randomized trial. J Clin Oncol 2002;20:2045–52. [Google Scholar] |
| 95. | Atkins MB, Hsu J, Lee S, Cohen GI, Flaherty LE, Sosman JA, et al. Phase III trial comparing concurrent biochemotherapy with cisplatin, vinblastine, dacarbazine, interleukin-2, and interferon alfa-2b with cisplatin, vinblastine, and dacarbazine alone in patients with metastatic malignant melanoma (E3695): A trial coordinated by the Eastern Cooperative Oncology Group. J Clin Oncol 2008;26:5748–54. [Google Scholar] |
| 96. | Keilholz U, Punt CJ, Gore M, Kruit W, Patel P, Lienard D, et al. Dacarbazine, cisplatin, and interferon-alfa-2b with or without interleukin-2 in metastatic melanoma: A randomized phase III trial (18951) of the European Organisation for Research and Treatment of Cancer Melanoma Group. J Clin Oncol 2005;23: 6747–55. [Google Scholar] |
Fulltext Views
2,332
PDF downloads
356



