
Translate this page into:
Spastic quadriparesis in Fabry disease: A diagnostic challenge
[To cite: Jha S, Vengalil S, Ravindranadh CM, Nalini A, Yadav R. Spastic quadriparesis in Fabry disease: A diagnostic challenge. Natl Med J India 2023;36:340–1. DOI: 10.25259/NMJI_MS_133_21]
Fabry disease (FD) is an X-linked inherited lysosomal storage disease caused by a defect in the GLA gene, which encodes the lysosomal enzyme alpha-galactosidase A resulting in the accumulation of glycosphingolipids—mainly globotriaosylceramide—in cells throughout the body, causing symptoms affecting multiple organs. Neurological symptoms in FD are most common and are the initial manifestations of FD. Small fibre neuropathy, premature strokes, autonomic dysfunction and large vessel dolichoectasia, and progressive sensorineural hearing loss are common manifestations, which generally appear early in the course of the disease.1 A family in England has been reported to have members affected with both FD and spastic paraplegia.2 We report progressive spastic quadriparesis as a presenting feature of FD, which must be considered a differential diagnosis in young individuals since the progression can now be retarded with early initiation of enzyme replacement therapy.
A 40-year-old shopkeeper, born out of non-consanguineous parentage, presented with insidious onset, gradually progressive spastic weakness of both lower limbs for 8 years, associated with dysphagia and dysarthria for 5 years without any sensory or bowel, bladder disturbances. He also had a history of painless, normoaesthetic, erythematous skin lesions since he was 20 years old, over the entire body including palms and soles, for which he was never evaluated. He did not have any addictions or any chronic drug intake. A detailed family history did not reveal any similar medical illness or other major medical illnesses in the family members. On examination, he was normotensive with bilateral pitting pedal oedema over the lower limbs and blanching erythematous maculopapular skin lesions over the entire body (Fig. 1). Neurological examination revealed spastic dysarthria, slow tongue movements, bilateral wasting of small muscles of the hand, grade 3 Modified Ashworth scale spasticity of all four limbs, with brisk deep tendon reflexes and bilateral plantars extensor. Formal power testing revealed grade 4/5 Modified Research Council power in bilateral shoulders, elbows and wrists group of muscles. Small muscles of the hands were weak and ankle dorsiflexors were bilaterally 1/5 with other lower limb muscles normal. He walked with a bilateral foot drop gait.

- Diffuse erythematous maculopapular lesions are seen over the back, shoulder and extensor aspect of the arm
Inflammatory and heredodegenerative causes of this spastic quadriparesis were planned to be ruled out. Investigations revealed microcytic hypochromic anaemia and albuminuria on spot urine analysis with fluctuating serum creatinine levels of 1.5–3 mg/dl (normal range 0.8–1.2 mg/dl); 24-hour protein excretion, vitamin B12, homocysteine, anti-nuclear antibody profile, serum copper, serum folate, liver function test, blood sugars were negative/normal. Magnetic resonance imaging (MRI) of the brain and spine showed few discrete and early confluent T2 FLAIR hyperintense white matter changes in bilateral parietal periventricular areas with normal angiogram of intracranial and extracranial vessels. Ultrasound abdomen and pelvis showed mild splenomegaly and 2D echocardiography with electrocardiogram was normal. Corneal biomicroscopy showed cornea verticillata and the skin lesions were angiokeratoma corporis diffusum on skin biopsy. Electromyography of the tibialis anterior and abductor digiti minimi of the right upper limb were sampled, which showed changes suggestive of chronic denervation. The sural nerve in the lower limb showed reduced sensory nerve action potentials. The median and ulnar nerves were sampled in the upper limbs, which were normal. Visual-evoked potentials and brainstem evoked auditory responses were normal. A diagnosis of FD was then thought of, which was confirmed by abnormally low leucocyte activity of the lysosomal enzyme alpha-galactosidase A (8 nmol/mg/hour; normal value 24–63). The cause of the pedal oedema was attributed to his serially elevated serum creatinine levels suggestive of a nephropathy secondary to FD. He was advised enzyme replacement therapy (ERT) along with angiotensin-converting enzyme inhibitors, aspirin and was explained the probable prognosis. Worsening nephropathy was a concern before initiating ERT for which a nephrology consultation was planned. The patient was lost to follow-up after discharge from our facility for a nephrology consult.
CORRESPONDENCE
Our patient with FD presented with pure motor spastic quadriparesis associated with cornea verticillata, cutaneous angiokeratomas, anaemia and albuminuria. Although FD usually appears in childhood or adolescence, atypical affected men can rarely have a neurological presentation in adulthood.3 Our patient interestingly had onset of skin lesions at 20 years of age and did not have acroparesthesias or previous strokes and transient ischaemic attacks which are well-known manifestations of FD. MRI brain in our patient showed posterior parietal predominant periventricular white matter T2 FLAIR hyperintensities (Fig. 2) which are reported to be the most common neuro-radiological manifestation of FD.4 FD is known to be associated with peripheral neuropathy, but coexistent anterior cell involvement might explain wasting and weakness in this patient, an association rarely seen in FD.
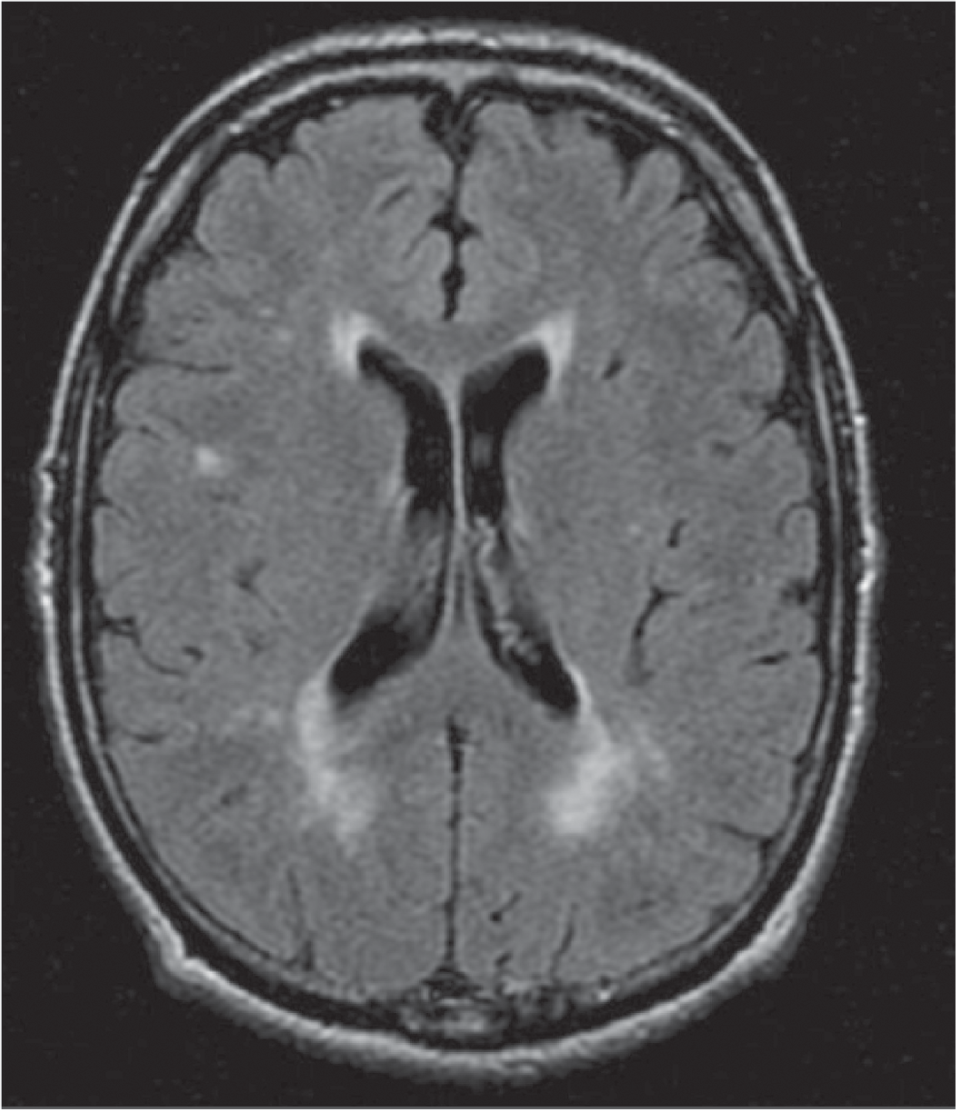
- Magnetic resonance imaging of the brain showing few discrete and early confluent T2 FLAIR hyperintense white matter changes in periventricular and posterior parietal white matter
The variability in its phenotypic expression and unpredictable clinical course make the diagnosis of FD challenging. Spastic quadriparesis is a rare neurological manifestation of FD and the diagnosis should be considered in a young man to offer ERT as a treatment option which is now available.
Conflicts of interest
None declared
References
- Fabry disease-Underestimated in the differential diagnosis of multiple sclerosis? PLoS One. 2013;8:e71894.
- [CrossRef] [PubMed] [Google Scholar]
- Study on a family with Anderson-Fabry's disease and associated familial spastic paraplegia. J Med Genet. 1976;13:455-61.
- [CrossRef] [PubMed] [Google Scholar]
- Neurological presentation of Fabry's disease in a 52 year old man. J Neurol Neurosurg Psychiatry. 2002;73:340-2.
- [CrossRef] [PubMed] [Google Scholar]
- CNS manifestations of Fabry's disease. Lancet Neurol. 2006;5:791-5.
- [CrossRef] [PubMed] [Google Scholar]



