
Translate this page into:
Unilateral retinitis pigmentosa associated with cystoid macular oedema
Correspondence to SAGNIK SEN; riksag@gmail.com
[To cite: Mishra C, Sen S, Kannan NB, Ramasamy K. Unilateral retinitis pigmentosa associated with cystoid macular oedema. Natl Med J India 2023;36:170–2. DOI: 10.25259/NMJI_651_20]
Abstract
Retinitis pigmentosa (RP) is the most common inherited cause of blindness in the developed world, characterized by night blindness, reduced central vision and constricted visual field; however, unilateral RP is extremely rare. Macular complications such as cystoid macular oedema (CME), macular holes and vitreoretinal interface alterations, such as epiretinal membranes, have been reported in advanced stages. We describe a patient with unilateral RP presenting with CME, a rare occurrence.
INTRODUCTION
Retinitis pigmentosa (RP) is the most common inherited cause of blindness in the developed world, characterized by night blindness, reduced central vision and a constricted visual field. Although the macula is usually spared from photoreceptor degeneration until advanced stages, cystoid macular oedema (CME), macular holes (MH) and vitreoretinal interface alterations, such as epiretinal membranes (ERM), have been reported.1,2 We describe a patient with unilateral RP, which is a rare form of the disease.
THE CASE
A 32-year-old woman presented to the retina clinic with complaints of diminution of vision in the left eye (LE) for the past 6 months. The patient’s first visit was 7 years ago, when her fundus examination revealed normal retina in the right eye (RE) and extensive mid-peripheral retinal bony spicule pigmentation in the LE, suggestive of RP with diminished full-field scotopic electroretinogram (ERG). The uncorrected visual acuity of the RE at presentation was 6/6 and LE was 6/60. With a distance correction of –1 DS/–2 DC at 165 degree, the visual acuity of the LE improved to 6/18. Near vision was measured as N6 in the RE and N24 in the LE. The anterior and posterior segment examination (Fig. 1a) in the RE were within normal limits, except for extreme peripheral retinal pigmentary change. The LE examination showed sluggish pupillary reactions, with mid-peripheral pigmentation in the retinal pigment epithelium (RPE), waxy pallor of optic disc and attenuation of blood vessels along with attenuated vessels (Fig. 1b) and a tubular central vision on confrontation testing. On ERG, grossly reduced rod and cone responses were observed in the LE (Fig. 1c). Autofluorescence imaging of RE macula was normal (Fig. 2a) and that of LE showed mid-peripheral hypofluorescence, and parafoveal ring-shaped zone of hyperfluorescence (Fig. 2b). Automated static perimetry showed normal fields in the RE (Fig. 2c) and constricted visual fields in the LE (Fig. 2d). Optical coherence tomography (OCT) through macula of RE was normal (Fig. 2e) and that of LE showed CME with thin ERM, subfoveal cystic changes along with suband para-foveal irregular RPE (Fig. 2f). The patient was diagnosed with unilateral RP with CME and advised topical dorzolamide 3 times daily. After a follow-up of 6 months, the CME remained unchanged.
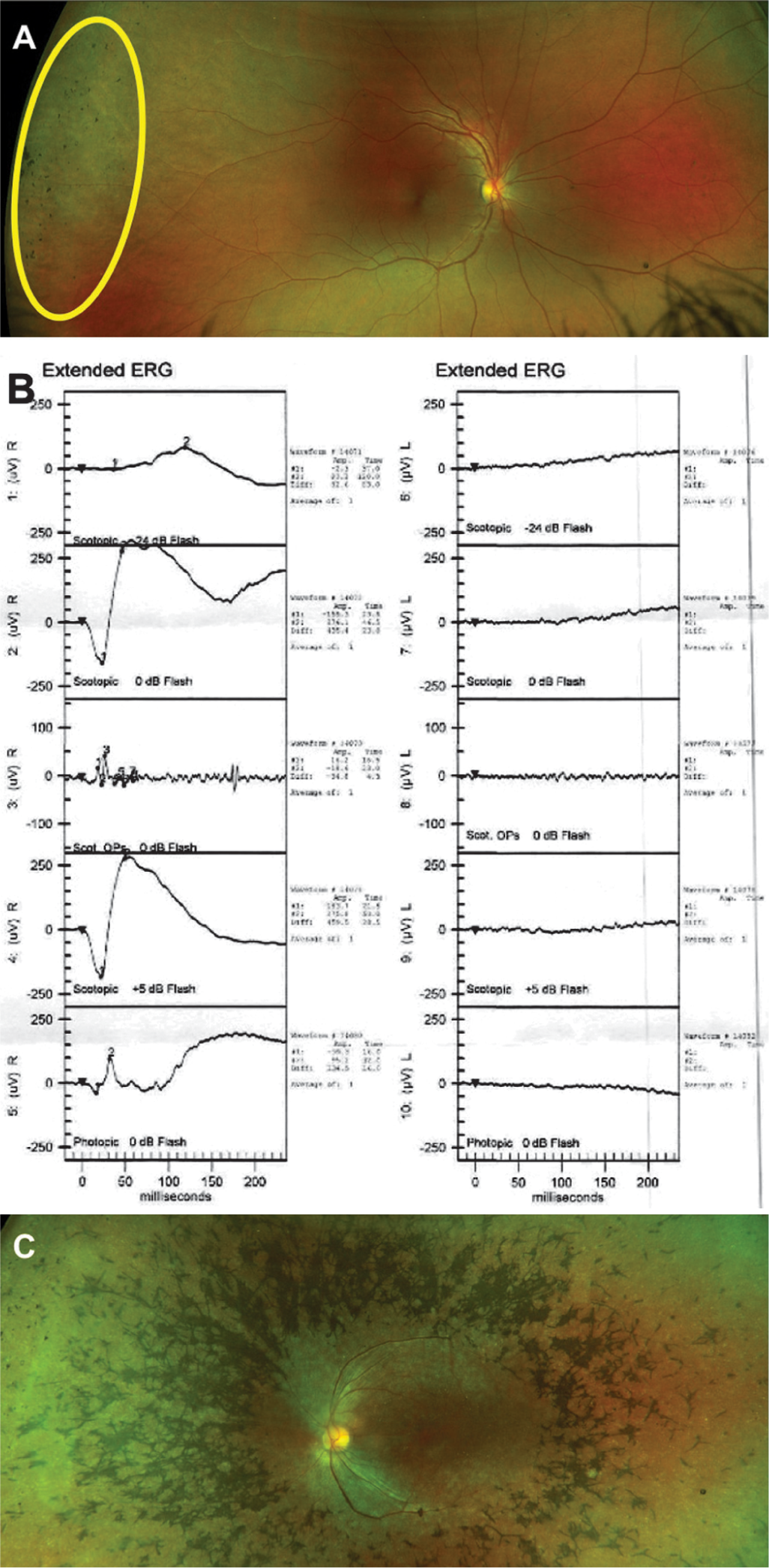
- Ultrawide pseudo-colour images of the fundus of the right eye (a) and the left eye (b). The right eye showed extreme peripheral retinal mild pigmentary change (highlighted in yellow), which was different from the usual location of pigmentary change characteristic of retinitis pigmentosa. The left eye shows extensive mid-peripheral retinal pigmentary abnormalities (bony spicules) sparing the macular region. (c) Electroretinogram of both eyes showing reduced scotopic amplitudes in the left eye
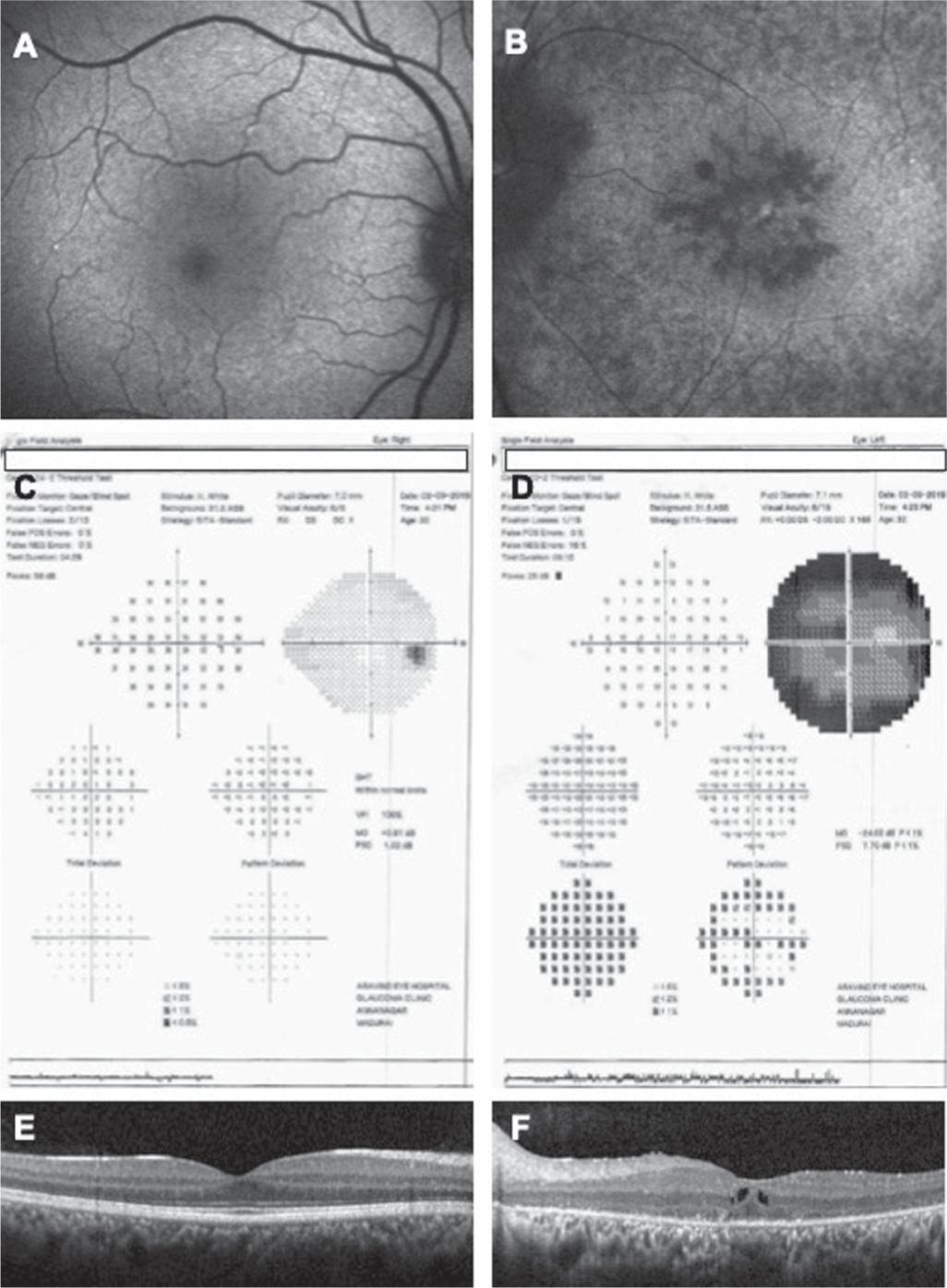
- Blue autofluorescence images of the right eye: (a) showing normal autofluorescence and the left eye (b) showing retinal pigment epithelium degeneration causing areas of hypofluorescence. (c and d) Automated static perimetry showing severely constricted visual fields in the left eye. Optical coherence tomography images of the right eye (e) showing normal fundus morphology and the left eye (f) showing increased central macular thickness with cystic spaces and loss of regularity in the ellipsoid zone
DISCUSSION
RP usually presents bilaterally. The incidence of unilateral RP has been reported to range between 0.2% and 5%.3 It may occur sporadically or in a hereditary (autosomal recessive, autosomal dominant, X-linked) pattern. However, unilateral RP is rarely associated with family history and presents much later than bilateral RP.4 Unilateral pathogenesis may be explained by the occurrence of genetic mosaics (mutation affecting cells selectively) or a somatic instead of a germline mutation.5 Unilateral RP may be associated with unilateral defective dark adaptation, deaf-mutism or mental retardation. However, the exact aetiology of unilateral RP is unknown.
Franceschetti et al. have defined the diagnostic criteria for unilateral RP as follows: (i) typical findings of RP in fundus of one eye; (ii) fellow eye showing normal fundus and full-field ERG; (iii) exclusion of differential diagnoses such as infectious, inflammatory and vascular causes for RP-like fundus lesions; and (iv) a minimum follow-up of 6 years.6 Potsidis et al. analysed the clinical course of 15 patients with unilateral RP and found that they had an annual decrease of 4.7% visual field area and 4.6% scotopic ERG amplitude. Moreover, this progression seemed to be faster in those who were younger. However, none of the published literature over the past century reported progression to involvement of the fellow eye, with one patient having a follow-up of 30 years.7 Because the apparently normal fellow eye compensates for the unilateral visual loss, patients tend to present late. However, we did not find any literature related to peripheral pigmentary changes in the fellow eyes; probably these changes might have been non-RP related, however the patient needs to be followed up for longer periods to confirm the same.
Several other aetiologies may be associated with a RP-like picture, e.g. retained metallic intraocular foreign body, infectious retinitis (syphilis, toxoplasma, rubella, measles), chronic inflammation, retinal drug toxicity, cancer-associated/auto-immune retinopathy, etc. Exclusion of these ‘phenocopies’ becomes more important if typical clinical appearance of RP is absent, e.g. absence of bone spicules. Infectious retinitis due to congenital infections (rubella, syphilis) may present unilaterally or bilaterally early on in life. However, the prognosis is better than that of RP. ERG may differentiate RP from infectious causes, with b-wave latency and amplitude both affected in RP, and unaffected latencies in syphilitic retinopathy. Moreover, infectious retinopathies usually do not show completely extinguished/unrecordable ERG as is commonly seen in RP.8,9
Our patient did not show any positive serology for infectious causes, and there was no history of psychiatric illnesses or intake of antipsychotic drugs.10 Following blunt trauma, pigmentary changes resembling RP occur due to RPE cell migration, but the progression pattern is not similar.11 Trauma was ruled out in our patient by a detailed history. Autoimmune retinopathies may be ruled out from RP by noting equally depreciated scotopic and photopic ERG, while in RP, scotopic ERG is affected earlier than photopic ERG.12,13 Long-standing posterior or intermediate uveitis can also hasten the pigmentation of retina, and in asymmetric cases they also need to be distinguished from unilateral RP.14 Our patient did not show any evidence of such chronic ocular inflammation or systemic diseases. A majority of patients initially reported as unilateral RP are finally diagnosed to have any one of these phenocopies. The importance of our report is that we have sufficient investigative back-up to confirm the diagnosis of unilateral RP.
CME in RP has been proposed to occur due to breakdown of inner and/or outer blood–retinal barrier, loss of polarized distribution of carbonic anhydrase IV in RPE cells and Müller cell swelling and dysfunction.15–18 CME may occur in 10%–50% of patients of RP.19 The occurrence of CME may not always be associated with loss of visual acuity, and depends mostly on the foveal thinning due to the primary pathology; however, CME is one of the treatable causes of central vision loss in patients with RP.19 Even after treatment, these eyes may show an anatomical improvement without any functional improvement, due to underlying tissue loss. Our patient had a considerably good visual acuity in spite of the presence of CME, in contrast to the other published patient of unilateral RP, who had a visual acuity of 3/60 in the presence of CME, probably because of advanced underlying foveal disease.20 Our patient was diagnosed with unilateral RP at a comparatively younger age. Moreover, she did not show anatomical or functional improvement of the CME, probably because the duration of follow-up was relatively small or because the CME was not in the active stage, and the cystic spaces probably reflected tissue loss due to underlying disease.
Several treatment methods have been described for RP-associated CME, e.g. topical carbonic anhydrase inhibitors, topical non-steroidal anti-inflammatory agents, steroids, etc.19 The majority of studies on treatment of CME in RP are isolated case reports or non-comparative retrospective trials, and based on these data, carbonic anhydrase inhibitors appear to be the preferable choice as first-line drugs for such eyes.19 We had put our patient on topical dorzolamide; however, at the final follow-up, the visual acuity and OCT features remained almost unchanged. Since the patient followed up with us for only 6 months, we did not try intravitreal interventions, etc. especially because the loss of visual acuity had not progressed considerably. The relevance of CME in our patient is that initially it can be mistaken to be due to the other RP phenocopies such as inflammatory diseases, which can also lead to CME on OCT. Hence, if proper differentiation is not done to rule out these phenocopies, the patient may end up being treated with multiple modalities targeting a completely different cause. This report is a reminder of such untoward circumstances, especially since patients with unilateral RP are adults and do not always give a family history or a typical history of night blindness.
In summary, unilateral RP is an uncommon disease entity, with less than 100 patients reported in the literature, and only three eyes reported from India, once in 1962 and then in 2018.21–23 Considering the rarity of this condition, we believe that our report adds value to the existing literature regarding this disease, and the supportive investigations will help readers to understand the workup of such eyes in detail.
Conflicts of interest
None declared
References
- Macular abnormalities in patients with retinitis pigmentosa: Prevalence on OCT examination and outcomes of vitreoretinal surgery. Acta Ophthalmol. 2011;89:e122-e125.
- [CrossRef] [PubMed] [Google Scholar]
- Macular lesions associated with retinitis pigmentosa. Arch Ophthalmol. 1977;95:798-803.
- [CrossRef] [PubMed] [Google Scholar]
- Unilateral retinitis pigmentosa and conerod dystrophy. Clin Ophthalmol. 2009;3:263-70.
- [CrossRef] [PubMed] [Google Scholar]
- Non-syphilitic unilateral retinitis pigmentosa. Am J Ophthalmol. 1968;65:573-4.
- [CrossRef] [PubMed] [Google Scholar]
- Unilateral retinitis pigmentosa: A proposal of genetic pathogenic mechanisms. Eur J Ophthalmol. 2012;22:654-60.
- [CrossRef] [PubMed] [Google Scholar]
- Chorioretinal heredodegenerations: An updated report of La Société d'ophthalmologie. In: Springfield. IL: Charles C Thomas; 1974. p. :266-74.
- [Google Scholar]
- Disease course of patients with unilateral pigmentary retinopathy. Invest Ophthalmol Vis Sci. 2011;52:9244-9.
- [CrossRef] [PubMed] [Google Scholar]
- Images in clinical medicine, Salt-and-pepper retinopathy of rubella. N Engl J Med. 2006;355:499.
- [CrossRef] [PubMed] [Google Scholar]
- Pseudoretinitis pigmentosa due to sub-optimal treatment of neurosyphilis. Eye (Lond). 1996;10:759-60.
- [CrossRef] [PubMed] [Google Scholar]
- Pigmentary retinopathy associated with low-dose thioridazine treatment. Can Med Assoc J. 1985;132:737.
- [Google Scholar]
- Traumatic pigmentary retinopathy. Am J Ophthalmol. 1981;92:621-4.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical and immunological characterization of paraneoplastic retinopathy. Invest Ophthalmol Vis Sci. 2013;54:5424-31.
- [CrossRef] [PubMed] [Google Scholar]
- Hyperautofluorescent ring in autoimmune retinopathy. Retina. 2012;32:1385-94.
- [CrossRef] [PubMed] [Google Scholar]
- Progression of retinal pigmentation mimicking unilateral retinitis pigmentosa after bilateral pars planitis: A case report. BMC Ophthalmol. 2018;18:242.
- [CrossRef] [PubMed] [Google Scholar]
- Blood-retinal barrier breakdown in retinitis pigmentosa: Light and electron microscopic immunolocalization. Histol Histopathol. 1995;10:913-23.
- [Google Scholar]
- Immunological studies in retinitis pigmentosa associated with retinal vascular leakage. Br J Ophthalmol. 1978;62:183-7.
- [CrossRef] [PubMed] [Google Scholar]
- The retina and its disorders In: Breakdown of the blood-retinal barrier. Oxford: Elsevier; 2010. p. :52.
- [CrossRef] [Google Scholar]
- Müller cells as players in retinal degeneration and edema. Graefes Arch Clin Exp Ophthalmol. 2007;245:627-36.
- [CrossRef] [PubMed] [Google Scholar]
- Retinitis pigmentosa-associated cystoid macular oedema: Pathogenesis and avenues of intervention. Br J Ophthalmol. 2017;101:31-7.
- [CrossRef] [PubMed] [Google Scholar]
- Unilateral retinitis pigmentosa: 30 years follow-up. BMJ Case Rep. 2014;2014:bcr2013202236.
- [CrossRef] [PubMed] [Google Scholar]
- Unilateral retinitis pigmentosa: Clinical and electrophysiological diagnosis. Can J Ophthalmol. 2018;53:e94-e97.
- [CrossRef] [PubMed] [Google Scholar]
- Unilateral retinitis pigmentosa. Clin Exp Optom. 2010;93:102-4.
- [CrossRef] [PubMed] [Google Scholar]



